








方案详情
文
我们借助于TSQ Altis的高灵敏度建立了一种基于沉淀酶解法的IgG-1类型单抗药物临床前生物分析的通用方法。该方法操作简单,价格低廉且具有高度通用性,可使用于临床前动物模型的所有生物基质。此外该方法还可以在测量体内药物浓度的同时监控单抗大分子药物的结构完整性。该方法广泛适用于药物开发的早期筛选以及药物的临床前研究阶段,提供注册必需的药代和药效动力学以及吸收/分布/代谢/排泄数据。该方法可为广大药企和CRO公司节约大量的方法开发时间, 并提供更多维的信息帮助制药工作者在早期判断候选抗体的成药可能性。
方案详情

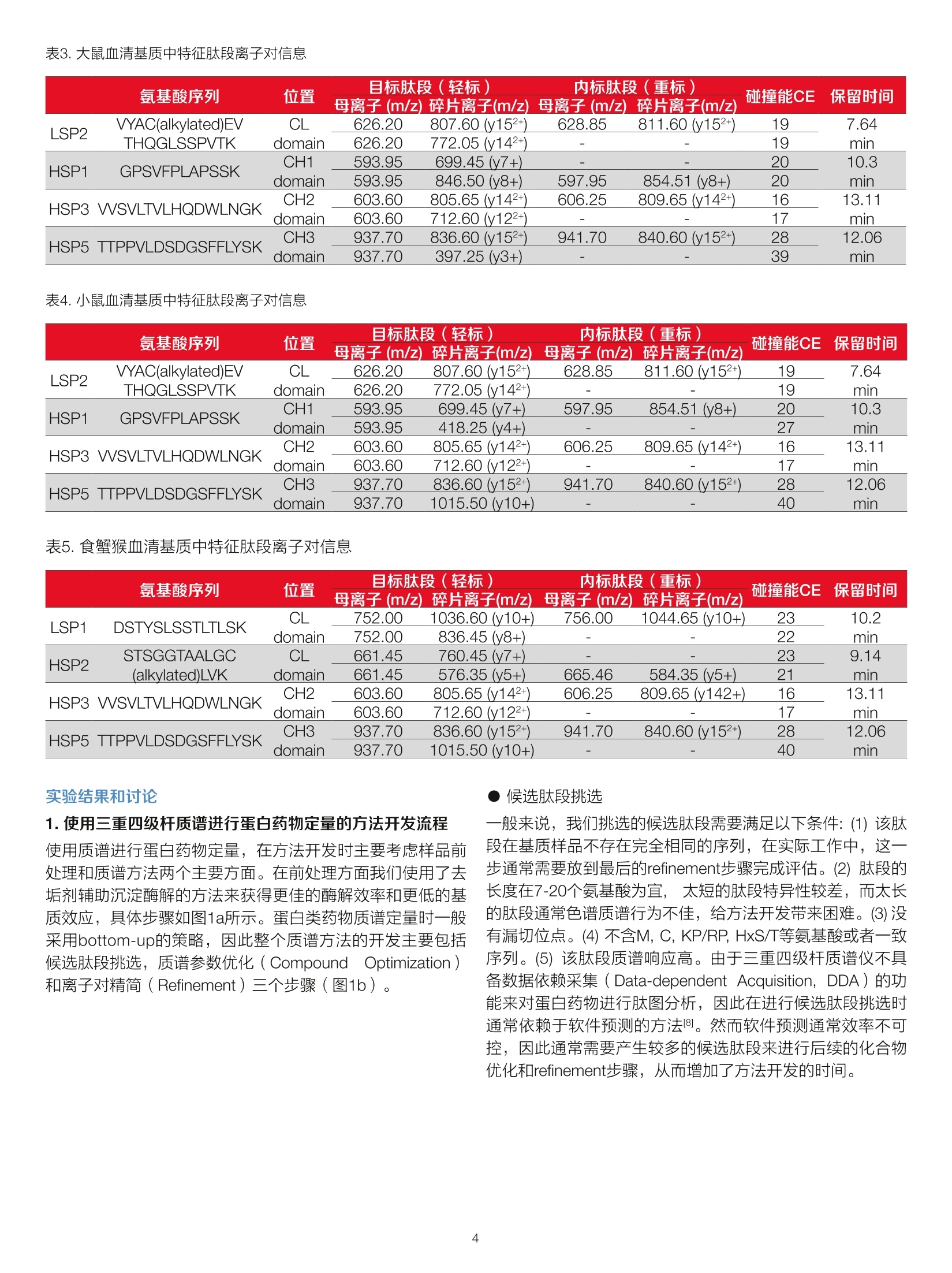
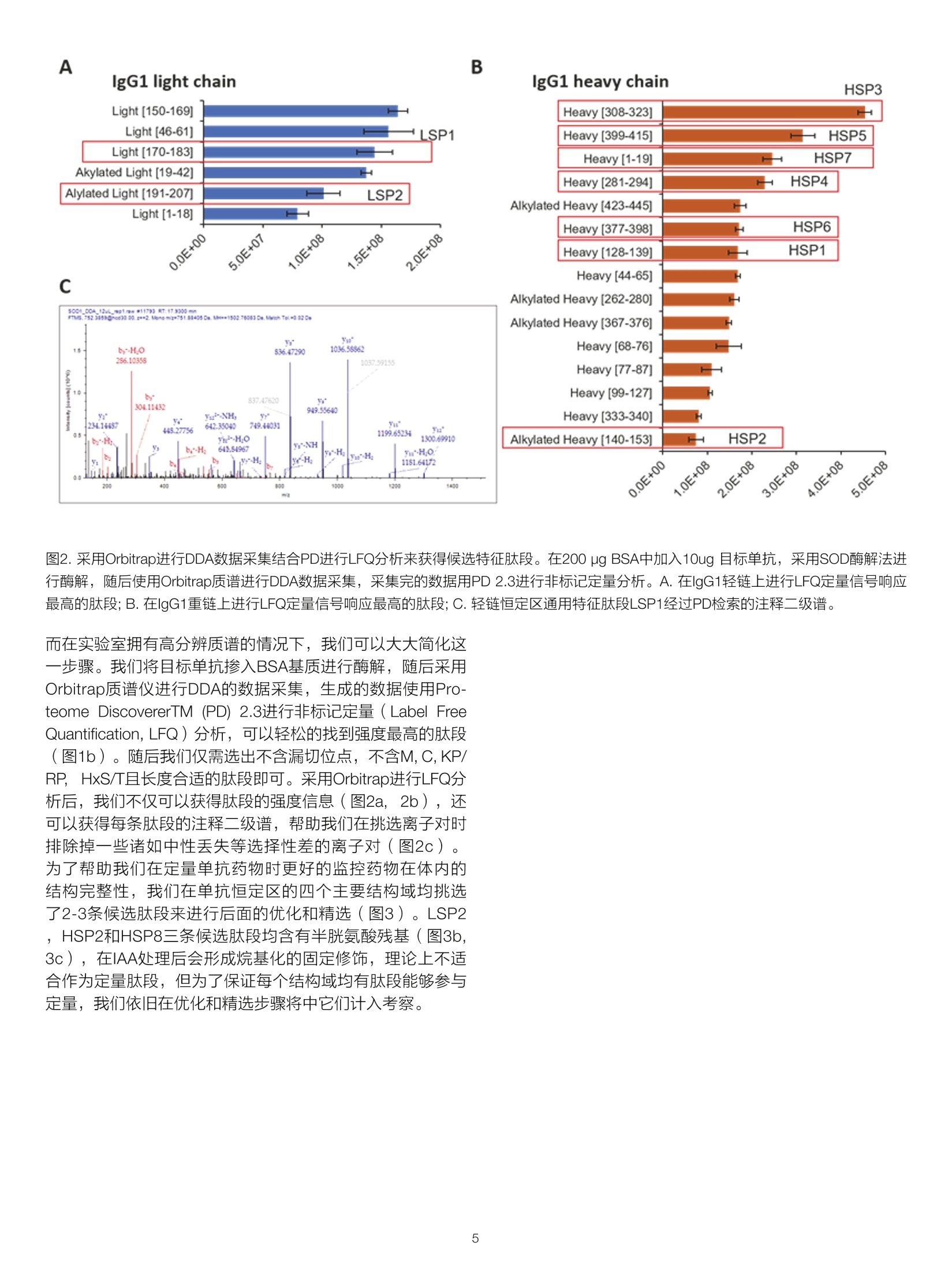
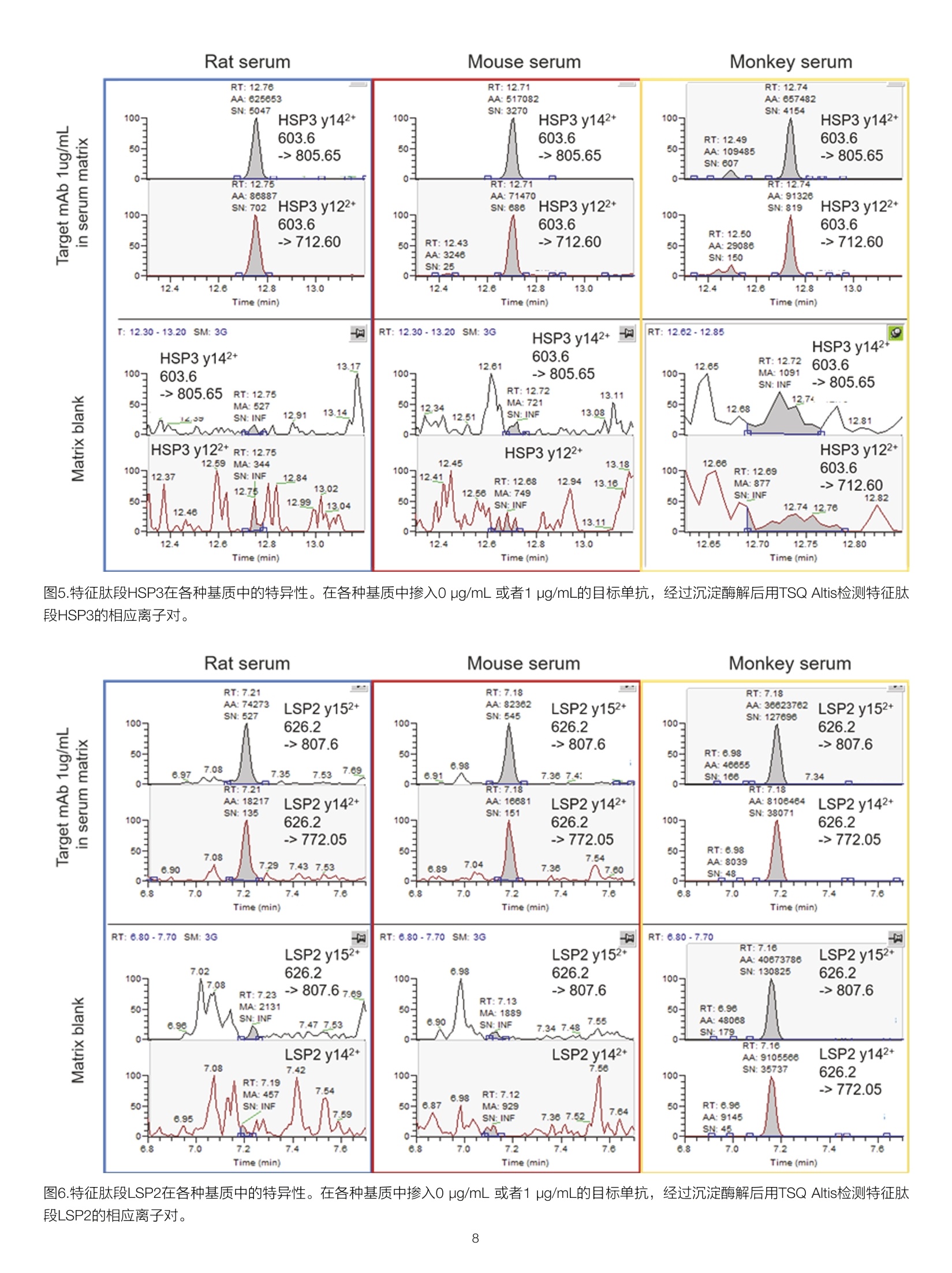
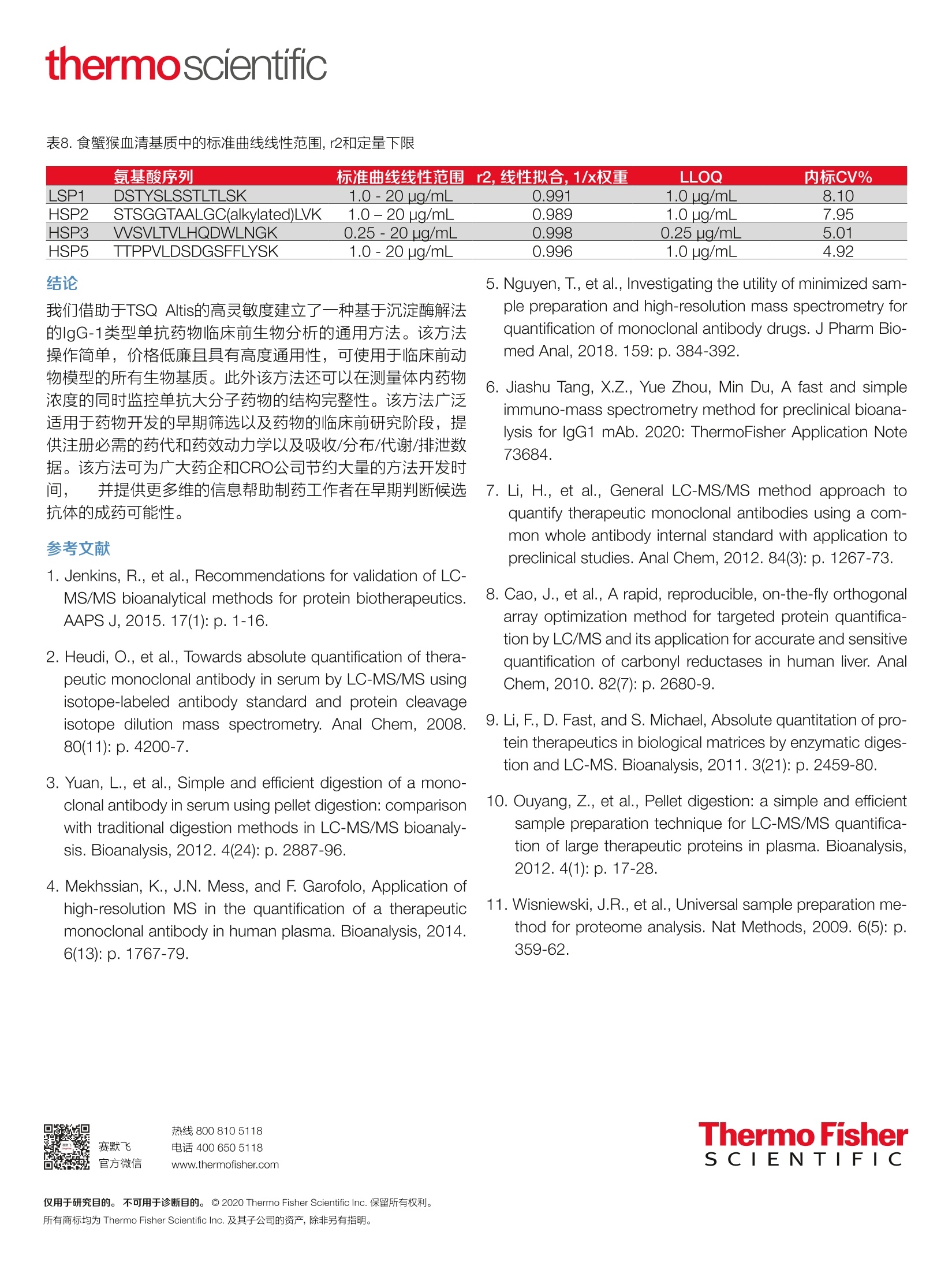
thermoscientific 样品前处理步骤 基于沉淀酶解法的IgG-1类型单克隆抗体药物临床前生物分析的通用方法 赛默飞世尔科技(中国)有限公司上海中国201206 关键词 单抗药物生物分析,临床前阶段,通用方法,抗药抗体,TSQAltis 方法优势 ●方法通用性,适用于所有的IgG-1类型单克隆抗体药物在临床前研究阶段的所有动物基质(包括血清和组织) 结构完整性,在测定生物基质中单抗药物浓度的同时监测药物结构的完整性,获得更加全面的药物代谢信息 样品前处理简单,不需要进行固相萃取等操作,也不需要进行任何优化,成本低 用于测定生物基质内的总体药物浓度,不受抗药抗体( Anti-Drug Antibody, ADA) 的影响 前言 生物分析对于生物大分子药物在最初的研发,临床前研究乃至临床研究阶段都具有非常重要的意义。为了提供药代动力学(pharmacokinetics, PK),药效动力学(pharmacodyna-mics, PD)和毒动学 (toxicokinetics, TK)所需的时间序列数据,我们对在生物基质中测定生物大分子药物浓度的准确性,灵敏度,选择性和通量均提出了非常高的要求求。基于免疫结合的方法(Ligand Binding Assay), 例如联免疫吸附(ELISA)方法由于其出色的灵敏度和通量,一直是生物大分子药物生物分析的金标准方法。但其固有的一些不足也在后续的研究中一直困扰着单抗药物的生物分析: (1)免疫法通常依赖于一对高质量抗体来识别蛋白药物的不同抗原表位。这些试剂其开发周期较长,在药物开发的早期通常难以获得。 (2)免疫法的线性范围较窄,在一些情况下不能完全符合药代动力学实验的需求。 (3)易受基质干扰影响,方法通常限定于特定的生物基质。 (4)易受动物体内抗药物抗体的影响。 和LBA方法不同,质谱法是基于目标蛋白特征肽段的氨基酸独特序列信息来实现方法的选择性,从而降低了对关键试剂的依赖。目前大多数由质谱进行的生物分析方法都采用了自下而上的蛋白质组学策略(bottom-up strategy),这种方法需要将蛋白质药物首先酶解成肽段,随后采用三重四级杆质谱进行选择反应监控(Selected Reaction Monitoring, SRM)[2,3]或者高分辨质谱进行平行反应监控(Parallel ReactionMonitoring, PRM) [4.5的方式来定量该蛋白药物的一条或者多条特征肽段。 对于治疗单抗药物建立一个通用的生物分析方法对于制药工作者来说具有非常大的意义,尤其对于在药物的早期研发阶段进行筛选时。介于目前在各药企研发管线上的在研单抗药物大多是全人源化的,这为在临床前阶段实现单抗药物生物分析的通用方法提供了可行性。通过选择人源化抗体恒定区的通用肽段,我们可以在非人源的生物基质中实现特异性的检测人源化抗体。在样品前处理方面,直接酶解法和免疫富集酶解法在不同的实验需求下被使用。免疫富集酶解法,即亲和质谱法可以提供低至10ng/mL级的定量下限I6,7],但是亲和质谱法一般只适用于血清(血浆)样本,且对抗药抗体并不能完全容忍。在这篇工作中,我们使用去垢剂辅助的沉 淀酶解法(surfactant-aided precipitation/on-pellet-digestion,SOD ) 来进行样品前处理。相比于溶液内酶解,沉淀酶解法可以在酶解前即将生物基质中的一些抑制肽段离子化的物质,例如磷脂等去除,从而避免了在酶解后再进行固相萃取等繁琐的操作。此外,去垢剂的引入不仅可以帮助蛋白变性取得更好的酶解回收率,还使得该方法更好的兼容动物组织样品。我们在目标单抗恒定区的每个主要结构域内均选择了一条通用肽段来进行目标单抗的定量,使得我们可以在定量的同时获得蛋白药物在体内的结构完整性信息。在内标的选择上,我们使用了通用型IgG-1kappa型重同位素标记的抗体作为内标,其恒定区的序列和人IgG-1类型抗体完全一致,可以帮助消除从样品前处理到质谱分析每一步的系统误差,得到更稳定的结果。通过TSQ Altis的高灵敏度,我们可以在不进行免疫富集的情况下获得血清中200 ng/mL 的定量 下限,基本满足抗体药物药代动力学分析的需求。该方法适用于所有临床前动物模型的所有样本,包括血清,血浆和组织,且不受抗药抗体的影响,可为药物开发,尤其是早期筛选节约宝贵的时间。 实验方法 样品前处理主要试剂耗材 SILuTMMab K1-Stable Isotope Labeled Universal Monoclo-nal Antibody Standard (SigmaAldrich, P/N: MSQC6) Sequencing Grade Modified Trypsin (Promega, P/N: V5117)DL-Dithiothreitol (SigmaAldrich, P/N: D9163) 标准曲线和内标工作液配置 将目标单抗(贝伐珠单抗)制剂配制在各种动物血清中,形成终浓度为0.1pg/uL的储备液,每个ep管分装50pL,在-80℃长期冻存。在每天实验前,将0.1pg/pL的储备液用 动物血清稀释至20ug/mL, 随后通过梯度稀释配制出浓度为0.2,0.4,0.625,, 1.25,2.5,5,10,20ug/mL的标准曲线。稳定同位素标记的SILumAb K1内标蛋白按照供应商的说明使用0.1%FA/H2O溶解至0.2pg/pL, 随后用1% BSA/PBS溶液稀释至终浓度为20 pg/mL,分装后在-80℃长期冻存。 内标掺入:取1mg总蛋白含量所对应的血清样品(或者标隹曲线),加入10uL 内标工作液,随后加入合适体积的PBS溶液,使得终体积为80uL。不同动物血清总蛋白含量有一定差别,对于大鼠来说对应为20pL血清,加入10uL内标工作液和50uL PBS溶液。稀释后的样品总蛋白农度为12.5 pg/pL, SILumAb K1内标蛋白对应血清原始浓度为10 ug/mL。 蛋白变性/还原烷基化:取16pL稀释后的样品(含有200pg总蛋白),加入84uL 1.2%SDS的PBS溶液, PBS溶液的pH值为8.0。加入2uL 500 mM DTT溶液 (DTT的终浓度为10 mM), 56℃还原30分钟,随后冷却至室温。加入6.5pL 500 mM IAA溶液 (IAA的终浓度为30 mM), 室温暗处反应30分钟。 蛋白沉淀:加入1倍体积预冷丙酮(108uL), 溶液出现浑浊。随后再加入5倍体积预冷丙酮(540uL),剧烈震荡,随后-20度放置3小时(或者过夜)。12,000g4度离心20分钟,移去上清。加入600 uL 预冷acetone : H2O=6:1溶液,剧烈震荡,随后12,000g4度离心20分钟,移去上清,室温干燥10 min。 ●目标单抗酶解:加入120 uL 50mM Tris/HCI 溶液 (pH 8.2),加入32 pL胰酶溶液(胰酶溶液的浓度为0.25 ug/pL, 用10mM 醋酸溶解),37度600rpm震荡酶解90分钟,沉淀可以全部溶解。 酶解终止:12,000g离心2 min, 取出135pL上清,加入15pL 10% FA/50% ACN以完全终止酶解反应。随后进样20-50 uL样品用于LC-MS/MS检测。 液相色谱条件 Thermo ScientificTM VanquishTM Binary Flex UHPLC 包含以下组件 ·System Base Vanquish Flex (P/N VF-S01-A) ·Binary Pump F (P/N VF-P10-A-01) ·Column Compartment H (P/N VH-C10-A) ·MS Connection Kit Vanquish (P/N 6720.0405) · Vanquish F Pumps 100 uL Mixer Set (P/N 6044.5100) ·Vanquish Split Sampler HT Sample Loop, 100 pL (P/N6850.1913) 分离条件 流动相A:0.1%甲酸/水;流动相B:0.1%甲酸/乙腈 色谱柱: Thermo ScientificTM HyperSIL GoldTM C18 1.9 pm,2.1mm * 100 mm (P/N: 25002-102130) 柱温:50℃, still air 表1.液相色谱梯度 Time (min) Flow rate Flow path %A %B 0.0 0.4 mL/min To waste 95 5 1.8 0.4 mL/min Main path 95 5 2.0 0.4 mL/min Main path 95 5 13.0 0.4 mL/min Main path 65 35 15.0 0.4 mL/min Main path 10 90 16.9 0.4 mL/min Main path 10 90 17.0 0.4mL/min Main path 95 5 20.0 0.4 mL/min Main path 95 5 质谱条件 仪器Thermo ScientificTM TSQ AltisTM三重四级杆质谱仪 表2.质谱参数 源参数 质谱设置 极性 正离子模式 SRM scan 喷雾电压 3800V 源内裂解 0V 鞘气压力 50 Arb Q1隔离窗口 0.7(FWHM) 辅助气压力 15 Arb Q3隔离窗口 0.7 (FWHM) 反吹气压力 0 Arb 碰撞气压力 2.0 mTorr 雾化温度 300℃ 循环时间 0.5 sec 离子传输管温度350℃ 数据模式 Centroid 氨基酸序列 位置 目标肽段(轻标) 母离子 (m/z) 碎片离子(m/z) 内标肽段(重标) 母离子 (m/z) 碎片离子(m/z) 碰撞能CE 保留时间 LSP2 VYAC(alkylated)EV CL 626.20 807.60 (y152+) 628.85 811.60 (y152+) 19 7.64 THQGLSSPVTK domain 626.20 772.05(y142+) 19 min HSP1 CH1 593.95 699.45 (y7+) - - 20 10.3 GPSVFPLAPSSK domain 593.95 846.50(y8+) 597.95 854.51(y8+) 20 min HSP3 VSVLTVLHQDWLNGK CH2 603.60 805.65 (y142+) 606.25 809.65 (y142+) 16 13.11 domain 603.60 712.60 (y122+) - - 17 min HSP5 TTPPVLDSDGSFFLYSK CH3 937.70 836.60 (y152+) 941.70 840.60(y152+) 28 12.06 domain 937.70 397.25 (y3+) - - 39 min 表4.小鼠血清基质中特征肽段离子对信息 氨基酸序列 位置 目标肽段(轻标) 内标肽段(重标) 碰撞能CE 份 保留时间 LSP2 母离子 (m/z) 碎片离子(m/z) 母离子(m/z) 碎片离子(m/z) VYAC(alkylated)EV CL 626.20 807.60 (y152+) 628.85 811.60(y152+) 19 7.64 THQGLSSPVTK domain 626.20 772.05 (y142+) - - 19 min HSP1 GPSVFPLAPSSK CH1 593.95 699.45 (y7+) 597.95 854.51 (y8+)_ 20 10.3 domain 593.95 418.25 (y4+) - 27 min HSP3 WSVLTVLHQDWLNGK CH2 603.60 805.65(y142+) 606.25 809.65 (y142+) 16 13.11 domain 603.60 712.60 (y122+) - 17 min HSP5 TTPPVLDSDGSFFLYSK CH3 937.70 836.60 (y152+) 941.70 840.60(y152+) 28 12.06 domain 937.70 1015.50(y10+) - 40 min 表5.食蟹猴血清基质中特征肽段离子对信息 氨基酸序列 位置 目标肽段(轻标) 母离子 (m/z) 碎片离子(m/z) 内标肽段(重标) 母离子 (m/z) 碎片离子(m/z) 碰撞能CE 保留时间 LSP1 DSTYSLSSTLTLSK CL 752.00 1036.60(y10+) 756.00 1044.65(y10+) 23 10.2 domain 752.00 836.45(y8+) - - 22 min STSGGTAALGC HSP2(alkylated)LVK CL 661.45 760.45(y7+_ - - 23 9.14 domain 661.45 576.35 (y5+) 665.46 584.35 (y5+) 21 min HSP3 WSVLTVLHQDWLNGK CH2 603.60 805.65 (y142+) 606.25 809.65 (y142+) 16 13.11 domain 603.60 712.60 (v122+) - 17 min HSP5 TTPPVLDSDGSFFLYSK CH3 937.70 836.60(y152+) 941.70 840.60 (y152+) 28 12.06 domain 937.70 1015.50 (y10+) 一 40 min 1.使用三重四级杆质谱进行蛋白药物定量的方法开发流程 使用质谱进行蛋白药物定量,在方法开发时主要考虑样品前处理和质谱方法两个主要方面。在前处理方面我们使用了去垢剂辅助沉淀酶解的方法来获得更佳的酶解效率和更低的基质效应,具体步骤如图1a所示。蛋白类药物质谱定量时一般采用bottom-up的策略,因此整个质谱方法的开发主要包括候选肽段挑选,质谱参数优化(Compound Optimization)和离子对精简(Refinement)三个步骤(图1b)。 ●候选肽段挑选 一般来说,我们挑选的候选肽段需要满足以下条件:(1)该肽段在基质样品不存在完全相同的序列,在实际工作中,这一步通常需要放到最后的refinement步骤完成评估。(2)肽段的长度在7-20个氨基酸为宜,太短的肽段特异性较差,而太长的肽段通常色谱质谱行为不佳,给方法开发带来困难。(3)没有漏切位点。(4)不含M, C, KP/RP, HxS/T等氨基酸或者一致序列。(5)该肽段质谱响应高。由于三重四级杆质谱仪不具备数据依赖采集(Data-dependent Acquisition, DDA)的功能来对蛋白药物进行肽图分析,因此在进行候选肽段挑选时通常依赖于软件预测的方法。然而软件预测通常效率不可控,因此通常需要产生较多的候选肽段来进行后续的化合物优化和refinement步骤,从而增加了方法开发的时间。 IgG1 heavy chain C 图2.采用Orbitrap进行DDA数据采集结合PD进行LFQ分析来获得候选特征肽段。在200 pg BSA中加入10ug目标单抗,采用SOD酶解法进行酶解,随后使用Orbitrap质谱进行DDA数据采集,采集完的数据用PD 2.3进行非标记定量分析。A. 在IgG1轻链上进行LFQ定量信号响应最高的肽段;B. 在IgG1重链上进行LFQ定量信号响应最高的肽段;C.轻链恒定区通用特征肽段LSP1经过PD检索的注释二级谱。 B mAb light chain sequence 图3.人源化IgG-1类型抗体的序列比对和特征肽段选择 在使用三重四级杆质谱对肽段进行定量时,我们通常采用SRM的扫描方式来监控特定的离子对(transition)。在最终的定量工作中每条肽段我们一般会选择一个定量离子(quantitative ion)和一个定性离子(confirming ion)来完成定量和定性。但在优化步骤时我们通常会选择至少4个离子对进行优化,以保证在refinement阶段后我们能得到两个具有特异性的离子对(图4a)。在进行优化时,我们所使用的样品为单抗药物经过了酶解和脱盐,使用注射器和三通直接喷入质谱,样品的浓度建议为0.2pg/pL蛋白酶解产物。 具体的优化参数主要包括源参数(共有参数),母离子价态, Slens RF,碰撞参数和质量参数等,这些参数则是化合物特异的(图4c)。由于肽段的分子量更大从而导致了其第二甚至第三同位素峰会相当高,因此在低分辨质谱的表现上会成为一个更宽的质谱峰(图4b),因此对母离子和碎片离子的质量进行优化可以进一步提升离子对的质谱响应来提升灵敏度。在为每个肽段挑选离子对时,我们建议使用三重四级杆质谱在多个碰撞能下对母离子进行产物离子扫描,结合PD给出的注释二级谱图信息来综合挑选响应最高,且被合理注释的碎片离子(图4b)。 编号 序列 位置 修饰 价态 M+0 M+1 M+2 SlensRF 碎片离子 碎片质量(Light) 母离子优 化质量 子离子优化质量 CE HSP3 SVLTVLHQDWLNG K Heavy.V308- K323 13+ 603.342 603.676 604.010 65 1 y14*(805.440-805.939) 603.60 805.65 16 2 y12(712.390-712.888) 603.60 712.60 17 3 y12*-NH3 (596.809-597.319) 603.60 597.55 14 4 y11²(655.848-656.349) 603.60 656.05 17 C B Global parameters Probeposition, spray voltage,sheathgas, Aux gas, Sweep gas, lon transfertube temp, Vaporizertemp Collision parameters Collision energy, CID gas pressure foreachtransition Mass optimization0ptimize precursor and fragmental ionmass for each transition 图4.肽段在三重四级杆质谱上的优化流程,每个肽段在refinement前挑选4个和理论质量匹配的离子进行优化。A. 特征肽段HSP3的离子对优化信息列表;B. 特征肽段HSP3的一级全扫描和不同碰撞能下的产物离子扫描;C.肽段在三重四级杆质谱上的逐步优化流程和所需要优化的项目。 肽段和离子对精选(Refinement) 该步骤的主要工作为在实际样品基质中挑选出满足特异性需求的特征肽段和对应的离子对,以简化在优化时得到的繁杂的离子对列表。由于三重四级杆是低分辨质谱,因此通过理论序列比对得到的特异肽段在进行实际样品分析时并不一定能满足特异性需求。因此空白基质样品与基质加标样品进行SRM的采集才是肽段和离子对精选的金标准。通过实际样品的分析我们发现肽段HSP3的离子对603.6->805.65 (y142+)和603.6-> 712.60 (y122+)在三种基质中均满足特异性的要 求(图5),然而肽段LSP2仅在小鼠和大鼠的血清基质中满足特异性需求,而在食蟹猴血清中存在很强的内源干扰(图6)。因此在食蟹猴血清样本中,我们选择了信号响应较低的LSP1来代表目标单抗的轻链恒定区(表5)。同样CH1结构域的代表肽段HSP1有着良好的色谱质谱行为,然而其在食蟹猴血清中不满足特异性的要求,因此我们只能选择信号强度低且含有半胱氨酸残基的HSP2肽段作为妥协来代表CH1结构域。最终经过refinement步骤,在三种动物血清基质中挑选的特征肽段和其对应的离子对信息详见表3,4,5。 图5.特征肽段HSP3在各种基质中的特异性。在各种在质中掺入0 ug/mL 或者1 pg/mL的目标单抗,经过沉淀酶解后用TSQ Altis检测特征肽段HSP3的相应离子对。 图6.特征肽段LSP2在各种基质中的特异性。在各种基质中掺入0 pg/mL 或者1 pg/mL的目标单抗,经过沉淀酶解后用TSQ Altis检测特征肽段LSP2的相应离子对。 2.基于SOD酶解方法的样品前处理流程 在以往的工作中,多种蛋白质酶解的方法:例如溶液内酶解[2.9,沉淀酶解[3,10]和滤膜辅助酶解(Filter Aided SamplePreparation, FASP)[11]均被用于单抗药物的生物分析。我们最终选择了沉淀酶解的工作流程,因为其在沉淀步骤即可去除包括磷脂在内的干扰物质,从而不需要在酶解后进行固相萃取的繁琐操作。而且这个方法不需要依赖昂贵的胰酶兼容去垢剂,使得样品分析极具经济性。我们在蛋白沉淀前使用SDS进行了蛋白的变性,并在随后进行了还原烷基化,这样使得蛋白在沉淀后更易复溶和酶解,从而得到更高的样品处理回收率。由于我们在选择特征肽段时考虑了使用包含半胱氨酸残基的肽段,因此我们对还原烷基化的稳定性,以及溶液pH环境对还原烷基化效率的影响进行了考察。结果发现,在pH 7.4, 8.0和8.5的PBS溶液环境中进行还原烷基化对于含有半胱氨酸的特征肽段LSP2, HSP2和HSP8的回收率并无影响,且这些肽段的信号响应稳定(图7),提示我们在实际工作中也可以选用这些肽段来进行目标单抗的定量。 图7.还原烷基化时的pH值对各特征肽段回收率的影响。含有20pg/mL目标单抗的大鼠血清样本采用沉淀酶解法酶解后,对以上肽段的离子对进行SRM扫描。在进行酶解时,选用三种pH 环境(pH7.4,8.0,8.5)进行还原烷基化,余余的步骤均一致。 3. IgG-1kappa通用型同位素内标的表现 在目标蛋白定量工作中,加入重同位素标记的蛋白质作为内标是一个好的选择,用于尽可能的消除整个前处理过程以及质谱分析时的系统误差。我们使用了SILuTMMab K1通用型重同位素标记单抗,其抗体恒定区序列和人IgG-1 kappa型高度保守(图3b, 3c),且其序列中的所有赖氨酸和精氨酸均被替换为重同位素标记的赖氨酸和精氨酸,在酶解后可以产生足够的质量差用于和目标单抗药物的肽段在质谱上进行区分。在三重四级杆质谱上考察内标的表现需要考察量两个问题,一是其序列中的重同位素氨基酸的同位素纯度是否足够,是否有对轻标肽段通道的干扰;;-二是各种动物基质中是否存在对内标的干扰信号。我们将掺入了最终工作浓度SI-LuMab K1的动物血清(10pg/mL)和空白基质进行比对,考察上述两个问题。结果显示在掺入了内标的样本中,轻标肽段通道的信号响应和空白基质中一致,表明该内标蛋白的同位素纯度足够,不存在对轻标肽段通道的干扰(图8)。而在考察第二个问题的时候,我们发现绝大多数的肽段,其基质中存在的对内标的干扰信号均小于内标响应的4%,提示这种干扰不足以影响定量(图8)。肽段HSP1的内标肽段的597.95 -> 854.51 (y8+)离子对通道虽然信号响应较高,但我们发现大鼠血清基质中对该离子对通道有较强的干扰,因此我们在大鼠血清基质中选择了597.95 ->854.51 (y8+)离子对通道作为肽段HSP1的内标通道(图8,表3)。对于肽段HSP2,我们发现食蟹猴血清基质对其内标通道的干扰达到了12%左右(图8,表5),因此在实际工作中,我们如果需要启用HSP2肽段的定量结果,那么建议将内标蛋白SILumAbK1的浓度提升至20 pg/mL。 图8.同位素内标在进行SRM定量时的表现。各种动物血清基质中加入10 pg/mL的SILumAb K1同位素内标蛋白,采用SOD酶解法进行酶解后检测重标特征肽段的离子对以及对应的非标记肽段的离子对。各种动物空白血清基质酶解后检测重标特征肽段的离子对,以观察在基质中是否存在对内标的干扰信号。 A 大鼠血清 C 小鼠血清 D 食蟹猴血清 图9.IgG-1通用定量肽段HSP3在各种动物血清基质中的线性。标准曲线浓度从0.2 (0.25)-20 pg/mL。 我们在不同基质中对方法的整体定量性能进行了考察。在不同的基质中,四条特征肽段均展示出了良好的线性(图9)。在大鼠和小鼠基质中,我们选用了相同的特征肽段,其中HSP3的信号响应最佳,其能达到0.2 pg/mL的定量下限,而其余三条特征肽段的定量下限则为0.5 ug/mL(表6,7)。食蟹猴基质中由于存在更强的基质干扰,在选择CL和CH1结构域的特征肽段时,我们选择了满足特异性,但信号强度低的肽段LSP1和HSP2作为妥协(表5)。由于更强的基质背景干扰,肽段HSP3的定量下限为0.25 pg/mL, 而其余三条特征肽段的定量下限则为1.0 pg/mL(表8)。该方法的灵敏度可以基本满足目前大多数情况下单抗药物药代和药效动力学的研究,若有更高的灵敏度需求,则可以使用我们的基于亲和质谱的通用方法。我们最终将HSP3定义为定量肽段(quantitative peptide)用于定量目标单抗的浓度,将其余三条肽段定义为监控肽段(monito-ring peptide) 用于监控目标单抗在体内的完整性。 表6.大鼠血清基质中的标准曲线线性范围,r?和定量下限 氨基酸序列 标准曲线线性范围 r,线性拟合,1/x权重 LLOQ 内标CV% _SP2VYAC(alkylated)EVTHQGLSSPVTK 0.5-20 g/mL 0.992 0.5 ug/mL 7.50 HSP1 GPSVFPLAPSSK 0.5-20 ug/mL 0.994 0.5 pg/mL_ 5.34 HSP3 VSVLTVLHQDWLNGK 0.2-20 ug/mL 0.999 0.2 ug/mL 3.51 HSP5 TTPPVLDSDGSFFLYSK 0.5-20ug/mL 0.995 0.5 ug/mL 6.54 表7.小鼠血清基质中的标准曲线线性范围,r和定量下限 L_SP2 HSP1 氨基酸序列 标准曲线线性范围 r,线性拟合,1/x权重 LLOQ 内标CV% VYAC(alkylated)EVTHQGLSSPVTK 0.5-20 ug/mL 0.991 0.5 ug/mL 5.12 GPSVFPLAPSSK 0.5-20 ug/mL 0.996 0.5ug/mL 4.76 HSP3 VSVLTVLHQDWLNGK 0.2-20pg/mL 0.999 0.2 ug/mL 4.11 HSP5 TTPPVLDSDGSFFLYSK 0.5-20 pg/mL 0.994 0.5 ug/mL 7.04 thermoscientific 表8.食蟹猴血清基质中的标准曲线线性范围,r2和定量下限 氨基酸序列 标准曲线线性范围 r2,线性拟合,1/x权重 LLOQ 内标CV% LSP1 DSTYSLSSTLTLSK 1.0-20 ug/mL 0.991 1.0 ug/mL 8.10 HSP2 STSGGTAALGC(alkylated)LVK 1.0-20 ug/mL 0.989 1.0 ug/mL 7.95 HSP3 VVSVLTVLHQDWLNGK 0.25-20 ug/mL 0.998 0.25 ug/mL 5.01 HSP5 TTPPVLDSDGSFFLYSK 1.0-20 ug/mL 0.996 1.0 ug/mL 4.92 我们借助于TSQ Altis的高灵敏度建立了一种基于沉淀酶解法的IgG-1类型单抗药物临床前生物分析的通用方法。该方法操作简单,价格低廉且具有高度通用性,可使用于临床前动物模型的所有生物基质。此外该方法还可以在测量体内药物浓度的同时监控单抗大分子药物的结构完整性。该方法广泛适用于药物开发的早期筛选以及药物的临床前研究阶段,提供注册必需的药代和药效动力学以及吸收/分布/代谢/排泄数据。该方法可为广大药企和CRO公司节约大量的方法开发时间, 并提供更多维的信息帮助制药工作者在早期判断候选抗体的成药可能性。 ( 1. J enkins, R ., et al. , Recommendations for validation of L C-MS/MS bi o analytical m e thods for pr o tein biotherapeutics.AAPS J, 2 015. 17(1): p.1-16. ) ( 2. Heudi,O . , et al., Towards absolute qu a ntification of thera-peutic monoclonal antibody in serum by LC-MS/MS usingisotope-labeled antibody standard and protein cleavageisotope dilution mass spectrometry . Anal Chem, 2008.80(11): p. 4200-7. ) ( 3. Yuan, L ., et al., Simple and efficient digestion o f a mono-clonal antibody in serum using pellet digestion: comparisonwith traditiona l digestion methods in LC-MS/MS bioanaly-sis. Bioanalysis, 2012.4(24): p . 2887-96. ) ( 4. Mekhssian, K., J .N. M ess, and F. Garofolo, A p plication o f high-resolution M S in t h e quantification of a th e rapeuticmonoclonal antibody in human plasma. Bioanalysis, 20 1 4.6(13): p. 1767-79. ) ( 5. Nguyen, T., et a l ., Investigating the utility of minimized sam-ple preparation and high-resolution mass s pectrometry forquantification of monoclonal antibody drugs. J Pharm Bio-med Anal, 2018.159: p. 384-392. ) ( 6. Jiashu Tang, X.Z., Yue Zhou, M in Du, A fast and s i mpleimmuno-mass spectrometry method for preclinical bi o ana-lysis for lgG1 mAb. 2 020: ThermoFisher Application No t e 73684. ) ( 7. Li, H., et al., General LC-MS/MS m ethod a p proach toquantify therapeutic monoclonal antibodies using a com-mon w h ole antibody in t ernal s t andard with application t o preclinical studies. Anal Chem, 2012.84(3): p . 1 2 67-73. ) ( 8. Cao, J., e t a l., A rapid, reproducible, on-the-fly orthogonalarray optimization method for targeted protein quantifica-tion by LC/MS and its application for accurate and sensitivequantification of carbonyl reductases in human liver. AnalChem, 2010.82(7): p.2680-9. ) ( 9. Li, F. , D . Fast, and S. Michael, Absolute quantitation of pro-tein therapeutics in biological matrices by enzymatic diges-tion and LC-MS. B ioanalysis, 2011. 3 (21): p. 2459-80. ) ( 10. Ouyang, Z., e t a l ., P ellet digestion: a simple and e f ficientsample p reparation technique for LC-MS/MS quantifica-tion of large therapeutic proteins i n plasma. Bioanalysis,2012.4(1): p. 17-28. ) ( 11. Wisniewski, J.R., et a l., U niversal sample preparation m e -thod f or proteome analysis. Nat M ethods, 2009.6(5 ) : p. 359-62. ) 1. 使用三重四级杆质谱进行蛋白药物定量的方法开发流程使用质谱进行蛋白药物定量,在方法开发时主要考虑样品前处理和质谱方法两个主要方面。在前处理方面我们使用了去垢剂辅助沉淀酶解的方法来获得更佳的酶解效率和更低的基质效应,具体步骤如图1a所示。蛋白类药物质谱定量时一般采用bottom-up的策略,因此整个质谱方法的开发主要包括候选肽段挑选,质谱参数优化(Compound Optimization)和离子对精简(Renement)三个步骤(图1b)。● 候选肽段挑选一般来说,我们挑选的候选肽段需要满足以下条件: (1) 该肽段在基质样品不存在完全相同的序列,在实际工作中,这一步通常需要放到最后的renement步骤完成评估。(2) 肽段的长度在7-20个氨基酸为宜, 太短的肽段特异性较差,而太长的肽段通常色谱质谱行为不佳,给方法开发带来困难。(3) 没有漏切位点。(4) 不含M, C, KP/RP, HxS/T等氨基酸或者一致序列。(5) 该肽段质谱响应高。由于三重四级杆质谱仪不具备数据依赖采集(Data-dependent Acquisition, DDA)的功能来对蛋白药物进行肽图分析,因此在进行候选肽段挑选时通常依赖于软件预测的方法[8]。然而软件预测通常效率不可控,因此通常需要产生较多的候选肽段来进行后续的化合物优化和renement步骤,从而增加了方法开发的时间。而在实验室拥有高分辨质谱的情况下,我们可以大大简化这一步骤。我们将目标单抗掺入BSA基质进行酶解,随后采用Orbitrap质谱仪进行DDA的数据采集,生成的数据使用ProteomeDiscovererTM (PD) 2.3进行非标记定量(Label Free Quanti_cation, LFQ)分析,可以轻松的找到强度最高的肽段(图1b)。随后我们仅需选出不含漏切位点,不含M, C, KP/RP, HxS/T且长度合适的肽段即可。采用Orbitrap进行LFQ分析后,我们不仅可以获得肽段的强度信息(图2a, 2b),还可以获得每条肽段的注释二级谱,帮助我们在挑选离子对时排除掉一些诸如中性丢失等选择性差的离子对(图2c)。为了帮助我们在定量单抗药物时更好的监控药物在体内的结构完整性,我们在单抗恒定区的四个主要结构域均挑选了2-3条候选肽段来进行后面的优化和精选(图3)。LSP2,HSP2和HSP8三条候选肽段均含有半胱氨酸残基(图3b,3c),在IAA处理后会形成烷基化的固定修饰,理论上不适合作为定量肽段,但为了保证每个结构域均有肽段能够参与定量,我们依旧在优化和精选步骤将中它们计入考察。
确定








还剩10页未读,是否继续阅读?

产品配置单

赛默飞世尔科技(中国)有限公司集团展位为您提供《生物基质中单抗药物检测方案(气质联用仪)》,该方案主要用于治疗类生物药品中生化检验检测,参考标准--,《生物基质中单抗药物检测方案(气质联用仪)》用到的仪器有赛默飞TSQ Duo 三重四极杆 GC-MS/MS
该厂商其他方案
更多