









在急性感染过程中,埃博拉病毒(EBOV)会引发从无症状表现到急性出血热等多种症状。而且,幸存者在无症状的恢复期也可能会传播病毒。在急性感染中,可能会发生细胞因子风暴(Cytokine Storm)和淋巴细胞凋亡,导致被感染者出现不可控的系统性炎症。近的研究证实,在感染过程中,埃博拉病毒蛋白VP40、糖蛋白(GP)和核蛋白(NP)会被装载到细胞外囊泡中。含有埃博拉病毒蛋白的细胞外囊泡已被证实能诱导受体免疫细胞凋亡,并且含有促炎性细胞因子。本篇文章中,研究者梳理了当前与埃博拉病毒相关的细胞外囊泡的研究现状,包括细胞外囊泡的生物发生机制、内含物以及它们对受体细胞的影响。另外,研究者讨论了埃博拉病毒所感染细胞产生的细胞外囊泡可能会引发的一些影响,提出了有待解决的问题和未来的研究方向。
方案详情

viruses Viruses 2019,11,4102 of 25 Viruses 2019, 11,410; doi:10.3390/v11050410www.mdpi.com/journal/viruses Review Extracellular Vesicles and Ebola Virus: A NewMechanism of Immune Evasion Michelle L. Pleet 1, Catherine DeMarino 1, Spencer W. Stonier 2,3D, John M. Dye 3,Steven Jacobson 4,M. Javad Aman 5D;) and Fatah Kashanchi 1* Laboratory of Molecular Virology, School of Systems Biology, George Mason University, Manassas, VA 20110,USA; mpleet@gmu.edu (M.L.P.); cdemarin@gmu.edu (C.D.) 2 Emergent BioSolutions, Gaithersburg, MD 20879, USA;stoniers@ebsi.com 3 Virology Division, U.S. Army Medical Research Institute of Infectious Diseases, Fort Detrick, Frederick,MD 21702, USA;john.m.dye1.civ@mail.mil 4 Viral Immunology Section, Neuroimmunology Branch, National Institute for Neurological Disease andStroke, National Institutes of Health, Bethesda, MD, 20892, USA; jacobsons@ninds.nih.gov 5 Integrated BioTherapeutics, Inc., Gaithersburg, MD 20850, USA;jaman@integratedbiotherapeutics.com * Correspondence: fkashanc@gmu.edu Received: 29 March 2019;Accepted: 1 May 2019; Published: 2 May 2019 check forupdates Abstract:IEbola virus (EBOV) disease can result in a range of symptoms anywhere fromvirtually asymptomatic to severe hemorrhagic fever during acute infection. Additionally,spans ofasymptomatic persistence in recovering survivors is possible, during which transmission of the virusmay occur. In acute infection, substantial cytokine storm and bystander lymphocyte apoptosis takeplace, resulting in uncontrolled, systemic inflammation in affected individuals. Recently,studieshave demonstrated the presence of EBOV proteins VP40, glycoprotein (GP), and nucleoprotein (NP)packaged into extracellular vesicles (EVs) during infection. EVs containing EBOV proteins have beenshown to induce apoptosis in recipient immune cells, as well as contain pro-inflammatory cytokines.In this manuscript, we review the current field of knowledge on EBOV EVs including the mechanismsof their biogenesis, their cargo and their effects in recipient cells. Furthermore, we discuss some of theeffects that may be induced by EBOV EVs that have not yet been characterized and highlight theremaining questions and future directions. Keywords: Ebola virus; exosome; extracellular vesicles; VP40; NP; GP;cytokine 1. Introduction 1.1. Ebola Virus Ebola virus (EBOV) is an enveloped, single-stranded RNA (-) virus known to cause severehemorrhagic fever in both humans and non-human primates (NHPs). Historically associated withsporadic outbreaks in Central Africa, EBOV more recently gained notoriety during the largest outbreakto date in West Africa (focused in Sierra Leone, Guinea, and Liberia), where the virus was responsiblefor >11,000 cases and >28,000 deaths [1]. At the conclusion of this outbreak in 2016, EBOV onceagain faded from the public spotlight; however, outbreaks have continued to occur, particularly in theDemocratic Republic of the Congo (DRC), where the virus is now considered endemic [2,3]. The EBOV genome is ~19 kb long and encodes seven open reading frames which translate toseven structural proteins. These structural proteins are the viral glycoprotein (GP1,2), nucleoprotein(NP), RNA-dependent RNA polymerase (L), and viral proteins 24 (VP24; minor matrix protein), VP30,VP35 and the major matrix protein VP40. Two additional non-structural proteins are also produced bytranscriptional processing of the GP mRNA-a soluble GP (sGP) and a small soluble GP (ssGP)[4-7]. It is estimated that only approximately 20%-25% of GP products are the membrane-anchored GP1,2form, while the remaining 75%-80% are of the secreted type, with ~70% sGP and ~5% ssGP [7-10].During early infection, EBOV targets mainly monocytes/macrophages and dendritic cells (DCs). It isthought that these migrating cells aid in systemic dissemination of the virus throughout the host,especially into secondary lymphoid organs and the liver. Intense replication of the virus then takes placein other host cell types, including endothelial cells, epithelial cells, fibroblasts and hepatocytes [11-14].Fatal filovirus infections often involve severely dysregulated innate and adaptive immune responses,defective coagulation, and are typified by systemic cytokine storm and multi-organ failure. Immunesystem damage includes type-I and type-II interferon (IFN) antagonism by VP35 and VP24, respectively,NK and T-cell depletion known as bystander lymphocyte apoptosis, and impaired DC maturation [15].Severe presentations of disease are accompanied by viral replication within and necrosis of the spleen,liver, kidneys, gonads, gastrointestinal tract and endocardium [13,16]. In those who are fortunate enough to survive acute Ebola virus disease (EVD), the virus becomesundetectable in the blood, depicting a typical phenotype of viral clearance. However, either entirevirus or viral components may remain detectable in patients relatively long after recovery, particularlyin immune-privileged sites [17-24]. Notably, recent studies have shown that over 1 in 4 male survivorsfrom the 2016 outbreak contained virus within their semen up to 7-9 months past the disappearanceof EVD symptoms. Furthermore, virus has still been detectable up to 16-18 months later [17]. Thistype of "clinical latency" or persistent infection has significance for public health in the followingways: (1) viral reservoirs within immune privileged sites have implications for reemergence andpersistence of the virus within the infected individual, potentially allowing for resurgence of infection,morbidity, and mortality [25]; (2) these reservoirs may allow for the transmission of the virus toadditional individuals long after initial infection, hindering efforts to control the spread of outbreaksby public health infrastructures [25]; and (3) significant political, socio-economic, and social stigmaconsiderations must be taken into account for policies to support the normal life and travel of EVDsurvivors [26-29]. Spread of the virus by this route is of particular concern, as several cases of sexualtransmission from male to female have already been documented.One of the first such cases involvedtransmission from a man 6 months past his recovery to a woman in Liberia [30,31]. To date, threeadditional "flare-ups" of EBOV by sexual contact have been determined, together accounting forapproximately half of the eight mini-outbreaks caused by persistently infected survivors documentedsince 2016 [32-35]. The most concerning of these cases occurred over 480 days (nearly 17 months) pastthe initial recovery of the patient [35]. To further complicate the matter, there have been multiple reportsaddressing potential asymptomatic or undiagnosed portions of the population. Previous studieshave put the number of seropositive but undiagnosed/asymptomatic individuals anywhere between0%-47%[36-44]. This of course implies that such individuals may represent an invisible source of viraltransmission and future outbreaks. Exemplifying this is a case involving an asymptomatic mother wholikely transmitted EBOV to her 9-month-old infant (as determined by sequencing and phylogeneticanalysis) through breast milk, ultimately resulting in the fatality of the child [19]. Interestingly, theasymptomatic father’s semen was also EBOV-positive, but was distantly related to the strains foundin the mother and child. Currently, the mechanism of this viral persistence is not well characterized,although new studies have hinted as several possibilities, which will be discussed later. 1.2. Extracellular Vesicles Extracellular vesicles (EVs) are small, membrane-bound vesicles that are released from numerouscell types and are involved in cell-to-cell communication. The intercellular communication is mediatedby EV cargo, consisting of nucleic acids and proteins, which can be transferred between cells toelicit a phenotypic change in the recipient cell [45-47]. EVs are heterogeneous; as such, the currentliterature separates EVs into several different categories based mainly on differences in size andcellular origin, including subtypes such as exosomes and microvesicles [48]. Microvesicles bud directlyfrom the plasma membrane and are generally larger than exosomes with diameters ranging from 100 to 1000 nm. Exosomes, which are formed from the fusion of multivesicular bodies (MVBs) withthe plasma membrane, are the smallest type of EV,and range approximately from 30 to 150 nm indiameter [49,50]. Exosomes are formed from the invagination of the endosomal membrane, leadingto the formation of intraluminal vesicles (ILVs) within the endosome, which is subsequently termedan MVB. During biogenesis, numerous soluble factors such as cytosolic proteins and nucleic acidsare captured and incorporated into these ILVs [45,46,49,50]. These components can be preferentiallypackaged through interactions with the endosomal sorting complexes required for transport (ESCRT)machinery. The ESCRT proteins form a series of cytosolic protein complexes described as the ESCRT-0,-I, -II, -III, and Vps4-Vta1 complexes [51,52]. These complexes are serially recruited to the endosometo form a pathway that recognizes, recruits and passes ubiquitinated cargo intended for disposal(i.e., through exocytosis or by degradation via lysosomal fusion) into nascent ILVs. Release of thispreferentially packaged cargo within exosomes can then take place upon fusion of the MVB with theplasma membrane. Although classical formation of ILVs and exosomes has been characterized to takeplace mainly via ESCRT-mediated packaging, alternative mechanisms of exosomal biogenesis havebeen demonstrated. This was initially observed through the generation of ILVs in cell lines despite thefunctional knockout of all four ESCRT complexes [53]. These proposed ESCRT-independent routesinclude CD63-mediated interactions [54,55], and by means of lipids such as neutral sphingomyelinase(nSMase; ceramide-dependent exosome formation) and phospholipase D2 (PLD2; syntenin-dependentexosome formation) [56,57]. Several Rab GTPases are also known to play important roles in intracellularvesicle transport, and thereby can participate in the biogenesis of exosomes through shuttling ofendosomes or Golgi vesicles to maturing MVBs [58]. Rab7 specifically has been shown to be involvedin these mechanisms [59,60], whereas Rab5, Rab7, Rab11, Rab35 and especially Rab27 have allbeen found to be involved in vesicular release from donor cells of various types [61-69]. However,ESCRT-independent mechanisms of exosomal biogenesis have been both far less characterized andconsidered less frequently, therefore the majority of exosome studies to date first focus on the ESCRTpathways for mechanistic studies. The selective incorporation of components into exosomes allows for unique exosomal profilesamongst cell types and can reflect differences in donor cell states, particularly during infection [70-72].Not surprisingly, several viruses have developed mechanisms to hijack exosomal-7 5b1i o1genesis andESCRT pathways/protein components to promote the viral life cycle and budding [73-75]. For example,the human immunodeficiency virus (HIV) Gag and the EBOV VP40 proteins have both been found tointeract with TSG101, a component of the ESCRT-I complex, and Alix, an accessory ESCRT pathwayprotein, to facilitate viral release from infected cells [76-85]. Likewise, numerous viral componentsfrom a multitude of infections have been discovered within exosomes and EVs, and have been shownto impact recipient cells and ultimately play a role in the course of pathogenesis [70-72]. For thisreason, there has been greater focus on the role of EVs during viral pathogenesis, which now representsa rapidly growing field of study for an expanding number of viruses and infectious diseases. The heterogeneity of EVs has forced the development of several purification techniques gearedtowards the isolation of distinct EV populations. The classic gold standard of EV isolation involvesa series of stepwise ultracentrifugation steps to sediment total EV populations, followed by densitygradient separation (i.e.,iodixanol or sucrose) to further divide EVs into distinct density fractions [86,87].Using this standardized protocol, separation of EVs from protein or protein-RNA aggregates andpotentially viral particles is made possible [87-89]. However, this method requires somewhat largesample volumes and therefore may have restricted utility due to the volume limitations associated within vivo studies. More recently, this protocol has been modified to implement various EV precipitationreagents such as ExoMAX (SBI) to overcome large sample volume limitations [90]. The study of EVs inthe context of viral infection requires precise isolation techniques in order to effectively remove anycontaminating virions or virus-like particles (VLPs) from the isolated EVs preps. The methods requiredfor virion removal are largely dictated by the overall size of the virion in question and will likelyvary between viruses of interest. For example, the Ebola virion is approximately 1-2 um in diameter, allowing for the vast majority of it to be removed from the supernatant using 1-2 passes through a0.22 um filter [91-96]. On the other hand, the HIV virion measures only 100 nm in diameter, a sizesimilar to that of exosomes, thereby requiring exclusion through density gradient separation [90,97].Additionally, investigations into EVs which originate from BSL-4 level pathogens, such as EBOV, haveled to the implementation of methods which utilize disposable size-exclusion columns, such as theqEV columns (IZON), which can be used with either samples concentrated by ExoMAX or on rawpatient samples to isolate vesicles [96]. The protocols designed to efficiently separate various virusesaway from EVs along with expected results are shown in Figure 1. B) D) Figure 1. Separation of extracellular vesicles (EVs) from virus for downstream analysis. An optimizeda workflow for the separation of EVs away from virus from a variety of backgrounds, including fromcell culture supernatants (A) and patient samples (B). Expected profiles of Ebola virus (EBOV) vs.exosomal proteins when extracellular components (i.e., EV-containing cell culture supernatants orbiofluids) are separated by size with qEV (IZON) columns, as outlined in the panel A workflow (C).Positive control virus-like particle (VLP) (containing VP40, nucleoprotein (NP) and GP) profiles areshown in lane 1, whereas a typical profile of EV-containing fractions of supernatants that were eitherunfiltered (top panel) or filtered through 0.22 um (bottom panel) from cells expressing VP40, NP andGP, followed by separation on qEV size exclusion columns are shown. Expected profiles of maturehuman immunodeficiency virus (HIV) virions (as indicated by p24 capsid protein) vs. exosomal markerproteins when cell culture supernatants are separated by density, such as by iodixanol or sucrosegradients, as outlined in the panel A workflow (D). Profiles demonstrated here are the expected resultsfor iodixanol fractions in increments of 1.2% from 6.0%-18.0%. Fractions where either virions or VLPsare known to elute or sediment are indicated by red boxes. Fractions containing only exosomes and noviral particles are enclosed by green boxes. Utilization of these techniques which allow for separation of EVs away from virus or VLPs hasallowed for significant research into the mechanisms by which EVs can promote viral infections. Ourlab has recently found that EBOV proteins such as VP40, GP and NP are packaged into EVs releasedfrom EBOV-infected cells [95,96,98]. Furthermore, EBOV VP40-associated EVs can elicit bystanderlymphocyte apoptosis in recipient, uninfected T-cells and monocytes, thereby potentially representinga novel mechanism contributing to the overall immune deregulation observed in EBOV-infectedindividuals [95,96,98]. In this manuscript, we review the current findings on EBOV-related EVs,potential contributions of EVs to EBOV pathogenesis and discuss important avenues of future study. 2. EVs andVP40 The EBOV VP40 protein functions mainly as the major matrix protein of the virus and is responsiblefor driving the budding and egress of new viral particles. Expression of VP40 within cells is alsoknown to result in the formation of VLPs, which is amplified by the co-expression of other viralproteins NP and/or GP, and appear virtually indistinguishable from infectious virions [91,99-104].Interestingly, recent studies have shown additional, non-structural functions of VP40 that may playpotentially important roles in EBOV pathogenesis. Once such additional role has been shown throughthe presence of EBOV VP40 within EVs [95,96]. Analysis into the mechanisms behind the packagingof VP40 into EVs, and exosomes in particular, has elucidated numerous interactions of VP40 withhost proteins and pathways including exosomal packaging, cell cycle and transcriptional regulation.The outcomes of these interactions have been shown to impact the host cell in a variety of ways,which ultimately have been hypothesized to aid the virus both in replication and spread duringinfection [95,96]. The connections of VP40 with the host cell to affect its packaging into EVs aresummarized here. 2.1. VP40 and the ESCRT Pathway The mechanism of VP40 packaging into exosomes has previously been shown to beESCRT-driven [95,96]. This is perhaps not surprising, as it has previously been revealed thatinteractions between the EBOV VP40 late domain and components of the ESCRT pathway such asTSG101, Alix and VPS4 takes place to aid in budding of virions and VLPs. TSG101, part of the ESCRT-1complex, has been demonstrated to aid in the budding of EBOV virions and VLPs through connectionswith the P(T/S)AP domain of VP40 [99]. This is very similar to the actions of TSG101 with matrix orassociated proteins of other viruses, such as HIV Gag, the open reading frame 3 protein of hepatitis Evirus, and the C protein of Nipah virus [78,79,105-107]. Alix is also known to arbitrate various cellularand viral protein interactions with the ESCRT machinery for both integration into ILVs and viralbudding [108,109]. Indeed, the EBOV VP40 YPx(n)L/I late domain motif has been confirmed to recruitAlix to aid in viral budding and egress independently of TSG101 [76]. Furthermore, the vacuolarprotein sorting 4 (VPS4) protein (responsible for the final stage of ILV formation and various membranescission events such as cytokinesis) has been previously implicated to play a role in the buddingof EBOV VLPs, as demonstrated by the inhibition of efficient VLP release with the use of a VPS4dominant-negative mutant [77,83]. In accordance with these previous works, we have demonstratedthat VP40 expression leads to changes in TSG101, Alix and VPS4 protein levels and forms. Theseintracellular changes correlated with expression differences of other exosome and EV-related proteinssuch as CD63 tetraspanin and Alix in the secreted EVs. Furthermore, overall changes in other ESCRTcomponents (i.e., EAP20 and EAP45 (ESCRT-II) and CHMP-6 (ESCRT-III)) have been observed incells expressing VP40 [95,96]. Together, these documented ESCRT protein changes were linked to thepackaging of VP40 into exosomes and EVs, implicating an ESCRT dependence for this occurrence. Similarly, although not directly an ESCRT-related protein, efficient release of VLPs has beenshown to be aided by the activity of the E3 ubiquitin ligase NEDD4 on the PPxY domain of VP40.This mechanism was shown to take place either at the exclusion of, or was enhanced by, thecoordinated activity of TSG101 [99,110]. In further support of this, interferon-stimulated gene 15 2.2.VP40 Influence on the Host Cell Cycle The host cell cycle has often been implicated in viral infections. Indeed, over the years numerousviruses have been shown to dysregulate the cell cycle of host cells in order to help in viral replication,spread, and/or latency. Examples of such viruses include HIV, influenza viruses, human T-cell leukemiavirus type 1 (HTLV-1), herpesviruses, human papillomaviruses, hepatitis B and C viruses, infectiousbronchitis virus and Adenoviruses [115-118]. Along these lines, we have also recently shown that cellcycle is altered in cells constitutively producing EBOV VP40. Specifically, VP40 production was shownto ultimately result in a promotion of cell cycle advancement and an overall increased growth ratein host cells [96]. This promotion of cell cycling was largely attributed to an increase in the amountsof cyclin D1 protein, which was directly correlated to the amounts of VP40 n nrrbeing produced withinthe cell. Levels of cyclin D proteins have been shown to be specifically manipulated by multipleviruses (such as HTLV-1, human neurotropic JC virus (JCV), human respiratory syncytial virus (HRSV),human papillomavirus, and Epstein-Barr virus) to aid in their life cycles [119-124]. Additionally,increased cyclin D1 levels have been linked in many cases to oncogenic phenotypes and acceleratedcell cycling [125-127]. The normal role of cyclin D1 during cell cycle is to bind to its appropriatecyclin-dependent kinase (cdk) partner, cdk2 or cdk4, to activate its kinase activity, and to phosphorylateRb to stimulate G1 to S phase cell cycle progression [125]. Therefore, upregulation of cyclin D1 as seenin VP40-producing cells is expected to result in increased growth, consistent with observations of thesecells. Upregulation of other cyclins in addition to D1, including cyclins E, A, and B1, was also observedin cells producing VP40 [96]; however, attention was focused on cyclin D1 as the D family of cyclinsare the first to be upregulated during the initiation of cell cycle from G0. Consequently, the fact that cyclin D1 is upregulated in cells producing VP40 led to a new set ofquestions regarding the mechanism of upregulation. Several previous studies have shown EBOV VP40present within the nucleus of infected cells transiently at early phases of infection [128-130]. Theseworks led to closer investigations of nuclear VP40's presence and role, particularly in relation to cyclinD1 upregulation. It was found that in stably transfected 293T cells, VP40 protein was indeed present inthe nucleus at fairly high levels [96]. Furthermore, nuclear VP40 was found to be bound frequently tothe cyclin D1 promoter, along with p300 and RNA Pol II at slightly elevated levels. Together, thesestudies potentially implicate VP40 as a recruiter of transcription factors or activator of transcriptionof various host genes that may aid in cellular replication and growth [96]. It would stand to reasonthat this would be beneficial for the virus, as it has been demonstrated that EBOV replicates better inactively dividing cells, whereas inhibition of cell cycle reduced the numbers of progeny virus [131].Therefore, utilization of VP40 to induce accelerated growth through transcriptional upregulation ofcyclin D, such as in epithelial cell types that continue to grow and divide, could represent a previouslyunappreciated mechanism of pathogenesis for EBOV. The ESCRT pathway's role in the release of VP40 exosomes was shown to be mediated in a cellcycle-dependent manner. Levels of ESCRT complex and exosomal marker proteins have been shownto be altered at different phases of the cell cycle, with lower levels present in quiescent or G0 cellscompared to those in G1/S or G2/M [96]. Accordingly, lower levels of these proteins were correlatedwith significantly decreased numbers of EVs at G0 in cells [96]. These trends were true for both normal293T cells and those constitutively expressing EBOV VP40, although cells expressing VP40 tendedto produce vesicles that were both larger in diameter (on average, as determined by the mode of thepopulation by ZetaView NTA analysis) and fewer in number as compared to 293T [96]. Therefore, themost EVs were produced from all cell types during G1/S and G2/M phases of the cell cycle, suggestingthat progression through cell cycle may enhance exosome and EV generation. Since VP40 increases therate of cell cycling, these results could also imply that VP40 can amplify the rate of EV biogenesis bythe same mechanism. The augmentation of VP40-containing EV release through cell cycle progressionby VP40 could have significant implications for recipient cells, and the immune status of the hostin general. 2.3. EV-Associated VP40 and Recipient Bystander Cell Effects As indicated above, during EBOV pathogenesis, a massive bystander lymphocyte apoptosistakes place which is particularly pronounced in fatal cases of EVD. Several potential mechanismsfor this have been proposed, including Fas/FasL and TNF-TRAIL interactions, impaired DC/T-cellinteractions, and nitric oxide (NO) or viral glycoprotein-induced apoptosis [15,132-134]. Recently,VP40-containing EVs have been proposed as an additional possible mechanism for the induction ofbystander lymphocyte apoptosis during EVD. This was evidenced by the induction of cell death ofboth T-cells and monocytes upon incubation with VP40 EVs [95,96,98]. This induction of cell deathwas shown to be through apoptosis, and the effect was shown to be cell type specific, as recipient293T cells did not react in the same manner. Indeed, 293T cells had improved viability when treatedwith VP40 EVs [96]. This stark contrast has interesting implications for how EVs from infected cellsmay impact pathogenesis within individuals. There is the potential for EVs containing VP40 fromEBOV-infected cells to travel to recipient immune cells and thereby reduce the pool of surveillingT-cells and monocytes, allowing for the virus to replicate unhindered. Should this mechanism ofimmune modulation by EVs from infected cells prove true, it may be even more relevant in cases ofrecurrent or persistent infections. Hypothetically, if VP40-containing EVs from infected cells are ableto increase the viability of recipient epithelial cells, this may allow these cells to be more permissiveto long-term infection. Contrastingly, the same VP40 EVs may be able to suppress the host immunesurveillance, thereby allowing any persistently infected cells to remain and hide, especially withinimmune privileged sites. This potential model supporting continued infections has been described atlength elsewhere [98]. 3. EVs and NP motif at the N-terminus, which may be a binding site for VP35 to inhibit NP-RNA interactions duringtranscription and replication [136,140,141]. The NP C-terminus is poorly conserved and shares nohomology with other viruses of the same order. Alternating o-helices and B-sheets allow this domainto act as a center for protein-protein interactions, particularly with VP35 or VP40 during nucleocapsidassembly and packaging into virions [140,142,143]. While the role of EBOV NP as an RNA-bindingprotein has been well characterized, we speculate that there may be additional roles for EBOV NP thathave yet to be uncovered and may be consequential for cells that may incorporate this protein into EVs. The nucleoproteins from Crimean-Congo hemorrhagic fever virus (CCHFV) and Lassa virus(LASV), two unrelated hemorrhagic fever viruses with high mortality rates, were recently describedto have nuclease functions in addition to their roles in nucleocapsid formation [144-146].Structuralanalyses of these two nucleoproteins revealed similarities to known nucleases and subsequent studiesrevealed both NPs to have nuclease functions. In the case of LASV NP,its dsRNA-specific exonucleasefunction was important for blocking the activation of type I IFN responses [145]. Previous structuralstudies of EBOV NP have focused on an N-terminal“core" domain [141] or C-terminal tail [142],leaving the region comprised of amino acids 405-641 relatively uncharacterized [141]. It would beinteresting to analyze the structure of this region to see if additional functions for EBOV NP can beimputed based on similarity with other protein domains. Regardless of the lack of a structure for thisregion, studies have shown that EBOVNP mutants that lack amino acids 451-600 or 451-739 inhibitminigenome-driven reporter gene expression in a dose-dependent manner [136]. EBOV NP lackingamino acids 451-600 or 506-739 failed to form properly-sized nucleocapsids [136,137], which takentogether indicates an important role for this region in the function of NP and nucleocapsid formation,but a specific role has yet to be revealed. Several lysine residues in EBOVNP were recently described to be acetylated by recombinant histoneacetyltransferases (HAT) in vitro. K513 and K617 were found to be acetylated by P300/CREB-bindingprotein (P300/CBP) and/or P300/CBP-associated factor (PCAF) [147]. These two acetylated lysineresidues reside within a region that is less conserved among members of the Ebola virus genus,although K513 is conserved between EBOV and Bundibugyo virus (BDBV) but not Sudan virus(SUDV) (USAMRIID; BLASTp unpublished observation). Furthermore, as discussed above, K513 andK617 fall in a region of EBOV NP that is not important for its RNA-binding function or associationwith other EBOV proteins. Regardless, acetylated lysine residues can function as binding sites forbromodomain-containing proteins and as such these residues in EBOV NP may be docking sites forother host proteins. Many bromodomain-containing proteins have chromatin remodeling and/ortranscription modification functions [148] and one such protein, Brd3, has a critical function in theelicitation of type I IFN production [149]. Infection of macrophages with Vesicular stomatitis virus(VSV) downregulated Brd3 expression, which in turn inhibited the ability of macrophages to produceIFNβ[149], demonstrating that Brd3 can be targeted in viral infections to limit type I IFN responses.It is therefore tempting to speculate that the acetylation of various lysine residues in EBOV NP may actas decoy docking sites for Brd3 to inhibit type I IFN responses. Brd3 is primarily a nuclear protein,but EBOV NP has been demonstrated to traffic to perinuclear areas in infected cells [129] whichmay give EBOV NP access to Brd3 prior to its nuclear translocation. A cellular interactome analysisof EBOV NP with cellular proteins identified SMYD3 [150], a histone methyltransferase, which isreported to recruit Brd4 to positively regulate transcription of target genes [151]. This interactome studysets a precedent for the interaction of EBOV NP with proteins involved in transcription regulationbut nonetheless also establishes a potentially interesting pattern of NP involvement with hithertounheralded cellular processes. While no direct IFN-antagonist function has been described based on structural studies of EBOVNP, EBOV NP has been implicated in IFN antagonism based on mutations that were acquired upon theadaptation of wild-type EBOV to a mouse-adapted variant. Mouse-adapted EBOV has mutations inNP and VP24 that render the virus lethal in wild type mice [152]. A recombinant virus bearing onlythe NP mutation, denoted"WT-NPMA,"found in mouse-adapted EBOV, demonstrates resistance to type I IFN when grown in murine macrophages treated with type I IFN [152]. WT-NPMA virus did notgrow as well when IFN was introduced prior to or after infection, indicating that if the IFN response isactivated in advance, any IFN antagonism is less effective. The WT-NPMA mutation in mouse-adaptedEBOV changed a serine to glycine,so why this mutation is so consequential is not understood sincelysine acetylation would not have been changed. Nevertheless, this demonstrates an involvement ofEBOV NP with type I IFNs. Though any direct IFN-antagonistic function of EBOV NP has yet to be described, the releaseof EVs containing EBOV NP from EBOV-infected cells presents a tantalizing possibility that suchEVs may carry a payload that can negatively regulate dose-dependent levels of type I IFN responsesin recipient cells. The number of NP molecules or indeed their monomeric or multimeric state inEVs has yet to be characterized, so it is unclear how effective such immunoregulation in uninfectedrecipient cells based on EBOV NP in EVs might be. Given that various EBOV proteins have diverseroles in immune evasion and suppression [135], any change in"type I IFN competency" in cellsneighboring an EBOV-infected cell may predispose them to infection in turn. EBOV NP in EVs maytherefore be an early immunosuppressive strike that helps predispose the infected host to develop afulminant infection. Alternatively, the role of any RNAs bound by EV-associated NP have yet to be elucidated, butif present may possibly affect recipient cells. As mentioned above, NP strongly associates with viralRNA as it is replicated in inclusion bodies within the cytoplasm. However, this affinity of RNA byNP has been shown to not be specific for viral RNA only [143,153]. Therefore, it is possible thatthe NP packaged into EVs may be bound to a multitude of host RNAs of various sizes and withfunctional characteristics. In the future, sequencing of the RNAs bound by EV-associated NP would beparticularly interesting to determine any potential effects these RNAs may have in recipient cells. 4. EVs and GP As was recently shown with NP, we have also demonstrated EBOV GP within EVs fromEBOV-infected cells [96]. Entry of filoviruses into cells is dependent upon the GP protein displayedon the viral surface. GP is translated from a single open reading frame as a GPo precursor. GPo isthen processed through the endoplasmic reticulum (ER) and Golgi apparatus, where it is cleaved byfurin-like proteases to produce two subunits connected by a disulfide bond: GP1 and GP2 [135]. GP1is an ectodomain responsible for interactions with cell receptors, while GP2 is the transmembranedomain and important for membrane fusion [9,154]. A trimer of GP1,2 molecules compose the matureGP protein. The N-terminal section of the GP1 subunit, also known as the core protein, interactswith GP2 and forms the head of the protein that interacts with receptors. The C-terminus of GPiis known as the mucin domain, is positioned on top of the GP2 head, and is heavily glycosylated,thus producing a 'glycan cap'[154-156]. GP can be processed differentially, however. As mentionedabove, only ~20%-25% of GP products are the membrane-anchored GP1,2 form, whereas the rest ofGP proteins are in dimeric secreted forms, with ~70% sGP and ~5% ssGP [7-10]. Furthermore, a shedform of GP1,2 (shed GP) is generated by a cleavage event at the cell membrane by host tumor necrosisfactor-a converting enzyme (TACE). All four of these GP forms contain the same first 295 residues ofthe N-terminus, although their C-termini differ, resulting in different functions. Only GP1,2 and shedGP possess the heavily glycosylated mucin-like domains which mask the GP epitopes [10]. Filoviral entry is accomplished through several steps: attachment, uptake and fusion. First, targetcell attachment is mediated by numerous possible nonspecific attachments between the GP1 glycancap and host cell surface proteins including primarily C-type lectin family members (i.e., hMGL,asialoglycoprotein, DC-SIGN, DC-SIGN(R), L-SIGN and L-selectin), but also may involve variousB1-integrins,Axl and TIM-1 and -4. These later proposed interactions may be even more nonspecificand could involve interactions with phosphatidyl-serine on the filoviral membrane [154,155]. Uptake ofEBOV is accomplished through a micropinocytosis-like mechanism, characterized by actin-dependentplasma membrane ruffling and blebbing to engulf bulk cargo and extracellular fluid [157,158]. Initiation of this uptake has been shown to be strictly GP-dependent, but the specific mechanism is not wellcharacterized [155,158]. The virus is then trafficked to late endosomes and/or lysosomes, as indicatedby co-localization first with Rab5-positive early endosomes and then Rab7/LAMP-1-positive lateendosomes [157]. Within endosomal vesicles of lowered pH and reducing conditions, proteolyticcleavage of GP1,2 is performed by host cysteine proteases cathepsin B and/or cathepsin L [154]. Thiscleavage event removes portions of the glycan cap and mucin-like domain of GP1 [154]. Once thehead portion of GP1 has been exposed, viral attachment to the Niemann-Pick C1 (NPC1) host cell lateendosomal/lysosomal membrane protein can take place. Due to the proposed specificity and importanceof NPC1 for filoviral entry, this receptor is a promising target for antiviral development [154,155,159].Following binding to NPC1, viral fusion and release of the nucleocapsid into the cytoplasm is initiatedby conformational changes in the GP2 subunit and fusion with the endosomal membrane [154,155,160]. l.Incorporation as Viral Uncoating on Entry Fusion and nucleocapsid release into the cytoplasm NP-Coated Exosomeswith VP40inside, GP12outside, andno NP 'Genome ~1-6 hours post-infection Il. Exosome Packaging MVB RNA- - bound NP Figure 2. Possible mechanisms of unconventional EBOV protein secretion. Three proposed mechanismsof packaging of EBOV proteins VP40, GP, and NP into EVs are illustrated along with hypothesizedtimes of release. (I) Incorporation as viral uncoated on entry: Entry of infectious virions or VLPsinto host cells through endosomes results in cleavage of the outer portions of GP1,2, binding to thehost receptor, and fusion with the endosomal membrane to release the viral nucleocapsid into thecytoplasm. Viral GP and VP40 proteins remaining within the endosome after nucleocapsid releasemay become packaged into exosomes as a result of inward budding of the endosomal membrane,resulting in extracellular release an estimated 1-6 h post-infection. (II) Exosome packaging: classicalloading of cargo into exosomes through endosomal sorting complexes required for transport (ESCRT)pathway (potentially ubiquitinated VP40; Ub-VP40), Golgi vesicle transport (GP1,2 and soluble GP(sGP)),and other mechanisms (NP). Resulting hypothesized exosomes would be released approximately24-30+ h post-infection (after viral translation of new protein components), and contain GP1,2 onthe surface, and VP40 and NP on the inside. The secretion of free sGP may also take place throughfusion of multivesicular bodies (MVBs) with the plasma membrane. (III) Microvesicle budding: Theoutward blebbing of microvesicles from the plasma membrane containing nontraditionally assembledEBOV components. GP12 is estimated to be oriented on the surface, with VP40 and NP on the inside.Microvesicles are estimated to be released approximately 24-30+ h post-infection. Additional details ofthese models are discussed in the text. Beyond the potential timing and mechanisms of integration of GP into EVs, there are significantfunctional possibilities for GP-studded EVs during pathogenesis. It has been observed by several groups that GP, either in membrane-bound GP1,2 or shed GP forms, is able to induce substantialimmune activation of monocytes, macrophages, and DCs, resulting in chemokine and cytokine secretionthrough TLR4 stimulation [167-169]. Treatment with sGP did not induce the same phenotype, and itwas determined that the glycosylation of the mucin-like domain was essential for this to occur [167].GP-TLR4 induction of cytokines and chemokines functioned to recruit CD11b+ macrophages andCD11c* DCs in vivo [168]. Moreover, GP-TLR4-stimulated cells were shown to potentially haveenhanced budding of VLPs [169]. Along these lines, a recent study demonstrated that shed GP-treatedmonocytes were triggered into differentiation through TLR4 activation [170]. Interestingly, previouswork has shown that monocytes are somewhat inefficiently infected, whereas macrophages and DCsare much more permissive to productive entry and replication of EBOV, and that uptake of EBOV bymonocytes stimulates their differentiation. Maturation of these cells in turn upregulates both the hostcell receptor NPC1 and cathepsin B, which presumably aids in viral entry, fusion, and release into thecytoplasm from the endosome [171]. It is therefore not surprising that shed GP-TLR4 induction ofuninfected monocyte differentiation also enhanced new EBOV infection and eventual cell death [170].Together, these studies imply that stimulation of the TLR4 pathway by GP1,2 or shed GP in myeloidcells potentially serves to not only prime them for new infection and differentiation, but also topromote cytokine release to recruit additional immune targets for the virus. Additional roles forGP in pathogenesis have also been described. It was found that shed GP was able to increase thevascular permeability of endothelial cells both directly and indirectly through cytokines (both pro-and anti-inflammatory) released from myeloid cells agitated by TLR4 binding [167]. Furthermore,glycosylated forms of GP have been shown to play roles in both the inhibition of natural killer (NK)cells [172] and potentially in inducing T-cell death through TLR4 binding [173]. Should glycosylatedforms of GP be present on EVs, it is possible that similar mechanisms to these could be at work. Lastly,secreted forms of GP have been widely speculated to play roles in immune evasion mainly throughacting as decoys for antibodies during the course of infection [174,175]. If GP proves to be exposed onthe surface of EVs, with either one or multiple epitopes present, it is logical to assume that these couldalso serve as decoys for the virus to aid in immune evasion. 5. Remaining Questions While the presence of EBOVVP40,NP, and GP within exosomes and/or EVs has been demonstrated,many questions still remain about the mechanisms of packaging and functional consequences of thesevesicles. Firstly, what are the forms of these viral proteins that are primarily found encapsulated withinor exposed upon the surface of EVs? Is there one main form or multiple for each of these proteins? Arethey found as monomers or as multimeric units, particularly in the case of VP40 and NP? What are thepost-translational modifications found on these proteins, and what are the effects enacted by thosemodifications? The surface has hardly been scratched on the numerous possibilities and implicationsthat exist. It is likely that in-depth proteomics studies will be necessary to fully appreciate the breadthutility of the forms of EV-associated EBOV proteins. Another important consideration is the additional cargo within EVs that has not yet beencharacterized. While VP40,NP, and GP have been found, what additional cargo may be specificallypackaged within EVs as a result of these viral proteins? It has not yet been determined whether EBOVpackages any of its four other proteins (VP24, VP30, VP35 and L) into EVs. VP30 and L are importantfor the translation of viral products, whereas VP24 and VP35 have significant pathogenic functions inIFN antagonism and impairment of signaling. Due to its size, it seems unlikely that L will be foundwithin EVs of <220 nm diameter. However, should VP24 or VP35 be found within EVs, these couldimpart substantial immune interference within recipient cells, particularly if they are potential hostcells for the virus. Outside of cargo from viral origin, host cell cargo is another important consideration. Previouswork has observed an upregulation of several cytokines within EVs from VP40-producing cells [96].Among the upregulated cytokines were TGF-B1, IL-15, MCP-1, and IFN-y-cytokines known to Further questions into the type of vesicle primarily utilized for EBOV protein secretion are alsowarranted. Initial experiments have focused on the role of exosomes due to the more developedliterature on characteristics and biogenesis of these vesicles; however, other important types of vesiclessuch as microvesicles or secretory autophagosomes may be equally or of greater importance to thepathogenesis and unconventional secretion of EBOV components. We consider three likely scenariosfor the incorporation of EBOV VP40, NP and GP into EVs for secretion. (1) The incorporation of viralproducts as a side-effect of viral uncoating in the early endosome upon entry into host cells. Aftermacropinocytosis, host cathepsins cleave the outer portion of the GP1 mucin-like domain to reveal thereceptor binding site, which then binds to host receptor NPC1, allowing fusion of the viral envelopewith the endosomal membrane and release of the nucleocapsid into the cytoplasm. Logically, VP40 andGP of the virion will still remain within the endosome after release of the genome into the cytoplasm. Should this endosome undergo inward budding afterwards, these components may then becomeintegrated into nascent ILVs, which may then become exosomes containing cleaved GP1,2 on the outsideand VP40 on the inside. (2) Classical packaging into exosomes via ESCRT-dependent mechanisms.In this scenario, VP40 (potentially targeted by ubiquitination) becomes packaged into maturing ILVsthrough the concerted effort of ESCRT complexes, whereas both membrane-bound GP1,2 and freesGP may become integrated into MVBs through Golgi vesicles. Cytosolic NP (potentially bound toRNA) may become packaged into exosomes through an as-of-yet uncharacterized mechanism. Finalbiogenesis of exosomes by this model will result in exosomes that have GP1,2 on the outside, and VP40and RNA-bound NP on the inside. Upon MVB fusion with the plasma membrane, sGP may also bereleased in a free form. (3) Budding of microvesicles from the plasma membrane. As VP40, GP, and NPare all well-characterized to associate and assemble at the plasma membrane, it stands to reason thatshould some factor during assembly be defective, or if the host cell directs the budding process, thatmicrovesicles may be released rather than the classical VLP. We hypothesize that these microvesicleswill vary widely in diameter, although they will also have the same orientations of packaged EBOVproteins as would be present during classical exosomal biogenesis. As a conceptual distinction, wetherefore propose that although these microvesicles may bud from the plasma membrane and containthe same assembly of viral proteins as VLPs, that they are both morphologically and functionallydistinct. We would suggest that the difference between VLPs and EBOV microvesicles or other EVs islargely due to the driving force of secretion and final morphology-VLPs are filamentous and buddingis driven by VP40, whereas EVs are roughly spherical and budding is driven by the host cell. Duringinfection, one or multiple combinations of these proposed pathways likely takes place, and the extentto which each, if any, contribute to the observed EVs generated in experimental models and in vivoremains to be determined. The three hypothesized mechanisms of unconventional EBOV proteinsecretion have been summarized in Figure 2. The type of vesicle(s) utilized by EBOV proteins to be released from infected cells may havesignificant ramifications not only for the additional cargo involved (of both host cell and virus origin),but also the timing of release in comparison to infectious virus or VLPs. As mentioned above, wepropose that viral components may be packaged into and released from infected cells in various EVsat time points earlier than the release of virions. Since virus release has been observed starting at~48 h [129], it stands to reason that, since the host cell is continually creating EVs, during the generationof viral proteins to be integrated into new virions, many of these components may be packaged intoEVs simultaneously. Should some viral components become packaged into exosomes during entry asproposed in mechanism #1 (Figure 2), these especially are likely to be released relatively quickly uponinfection, perhaps even before new viral translation begins. If EVs containing GP are released prior tovirion release, it is possible that they may be able to then act as a tool to recruit and differentiate/primefuture targets of the maturing virions. Alternatively, should EVs containing VP40 be released earlierthan new virus, they may be able to either wipe out sentinel immune cells that are susceptible, oraccelerate the cell cycling of recipient epithelial cells to act as future sites of infection. The pathogenic potential of EBOV EVs is not limited to acute disease. As described above, manycases of recurrent, asymptomatic or persistent infections have arisen. The ability of EBOV to remainclinically latent in immune-privileged sites has not yet been well characterized, but a few recent studieshave offered possible hints to this effect. One recent study showed that retinal pigment epithelialcells could potentially serve as reservoirs for productive EBOV infection, and that apoptotic signalingcascades were downregulated in these cells. Additionally, infected retinal cells were induced to releaseseveral pro-inflammatory cytokines and chemokines [196]. In asymptomatic NHPs, persistent EBOVinfection and replication along with systemic inflammation was found in monocyte/macrophage(CD68+) cells in multiple compartments including in the vitreous human and adjacent tissue, theepididymis, and the brain [197]. If retinal pigment epithelial cells or adapted CD68+ cells resident inimmune-privileged sites are able to sustain EBOV infection, perhaps through VP40 upregulation ofcell cycle or other transcriptional events that downregulate cell death, these cells could then release a variety of EVs containing multiple EBOV components, which can then either (1) recruit and/or primeneighboring cells for infection through differentiation or other mechanisms; (2) induce apoptosis insurveilling T-cells; (3) continually barrage neighboring or distant sites with inflammatory mediatorssuch as cytokines or chemokines; or (4) act as decoys for any GP-targeting antibodies produced in theconvalescent host. Ultimately, any or all of these mechanisms could potentially allow for the lingeringpresence of EBOV and prevent its complete clearance for prolonged timeframes. 6. Conclusions We have reviewed here the current evidence of EBOV VP40, GP and NP protein packaging intohost cell EVs during the course of infection, and their possible functional effects within infected hosts.The capacity of the EBOV protein-containing EVs to exacerbate EBOV pathogenesis may be many-foldin a variety of situations. Depending upon the type of donor cell, stage of infection, physiologicalsite, and recipient cell, EVs from infected cells could potentially aid in the spread of infection throughrecruitment and priming of new target host cells, downregulation and/or upregulation of systemicinflammation, and by decimation of the immune cell populations in general. These effects could beimportant not only in acute infection, but also during persistent or asymptomatic infections. Forexample, during acute infection, infected cell EVs containing EBOV proteins could potentially beresponsible for both the killing of bystander T-cells and the priming and recruitment of myeloid cells forsubsequent infection, thus allowing the virus to spread unchecked systemically. Alternatively, duringpersistent infection in immune privileged sites, infected cells may release low, consistent levels ofEBOV protein-associated EVs, which can then potentially travel to peripheral areas and simultaneouslydestroy surveilling immune populations and serve as immune decoys for any circulating antibodiesagainst the viral GP. Both of these possibilities would represent novel mechanisms which could allowfor the virus to effectively evade host immune responses to infection. As this is a relatively new field, there are many diverse avenues for future study remaining.In particular, identification of the full assortment of EBOV proteins and their forms/orientations thatmay be packaged into EVs, as well as the functional consequences of cells receiving those EVs is ofsignificant interest. Additionally,characterization of the subtype (i.e., exosome vs. microvesicle) andbiogenesis pathway of the EVs mainly responsible for aiding in pathogenesis of the virus will becomeimportant, particularly if potential therapeutics will be aimed at this newly identified area. While thereare a multitude of questions that remain to be addressed, the evidence thus far certainly shows that theeffects of EBOV protein-containing EVs are clearly separate from those enacted by VLPs or virions,and therefore these EBOV EVs likely play their own important role during pathogenesis. For thesereasons, it will likely become increasingly important for future investigations into the effects of varioustypes of EVs or VLPs to carefully utilize specific isolation procedures so as to not result in ambiguousor misleading findings. Author Contributions: M.P., C.D., and S.W.S. wrote the manuscript. Overall direction of the project and editingwas accomplished by J.M.D., S.J., M.J.A., and F.K. Funding: This research was funded by the National Institutes of Health(NIH), grant numbers AI078859, AI074410,AI127351-01, AI043894, and NS099029 to F.K, and R01AI132204 to M.J.A. ( Acknowledgments: We would like to thank all memb e rs of the Kashanchi lab, especially James Erickson, Maria Cowen, and Gwen Cox. ) ( Conflicts of Interest: The authors declare no conflict of interest. ) Disclaimer: The views and conclusions contained in this document are those of the authors and should not beinterpreted as necessarily representing the official policies, either expressed or implied, of the US Department ofthe Army, the US Department of Defense, the US Department of Health and Human Services, or of the institutionsand companies affiliated with the authors. ( References ) 1. WHO. Ebola Outbreak 2014-2015.Available online: http://www.who.int/csr/disease/ebola/en/ (accessed on7 January 2019). ( 2. Ebola. E bola Situation Reports: Democratic Republic of the Congo. Avai l able online: http: // w ww.wh o .int/ebol a /si t u a t i on-reports/drc-2018/en/ (accessed on 7 January 2019). ) ( 3. WHO. Ebola Virus Disease—Democratic Republic of the Congo. Available online: ht tp :/ /www.who.int/csr/don/ 2 5- j ul y- 2018-ebola - drc / en/ ( a ccessed on 1 A u gust 2018). ) ( 4. Rougeron, V.;Feldmann, H.; Grard, G.; Becker, S.; Leroy, E.M. Ebola and Marburg haemorrhagic fever. J. Clin.Virol. 2015, 64,111-119. [CrossRef ][ PubMed] ) ( 5. MuHlberger, E. Filovirus replication and transcription. Future Virol. 2007,2,205-215 .[CrossRe f] [Pu b Med ] ) ( 6. Sanchez, A .; K iley, M.P.; Holloway, B.P. ; Auperin, D.D. Sequence analysis of the Ebola virus genome: Organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 199 3 , 29, 215-240. [ CrossRef] ) ( 7. Volchkov, V.E.; Becker, S.; Volchkova, V.A.; Ternovoj, V.A.; Kotov, A.N.; Netesov, S.V.; Klenk, H.-D.GP mRNAof Ebola Virus Is Edited by the Ebola Virus Polymerase and by T7 and Vaccinia Virus Polymerases1. Virology1995,214,421-430. [ CrossRef] ) ( 9. Manicassamy, B.;Wang,J.;Ru m schlag, E.; Tymen, S.; Volchkova, V.; Volchkov, V.;Rong, L. Characterizationof Marburg virus glycoprotein in viral entry. Virology 2007,358,79-88. [C r o ssRef] [ P ubM e d] ) ( 10. Cook, J . D.; L ee, J.E. The Secret Life of Viral Entry Glycoproteins: Mo onlighting in Immune Evasi o n.PLoS P athog. 2013,9, e1003258. [CrossRef ] [ P ubM e d] ) ( 11. L . Geisbert, T .W.; Hensley, L.E.; Larsen, T.; Young, H.A.; Reed, D.S.; Geisbert, J.B.; Scott, D.P.;Kagan, E.;Jahrling, P.B.; Davis, K.J. Pathogenesis of Ebola Hemorrhagic Fever in Cynomolgus Macaques: Evidence thatDendritic Cells Are Early and S ustained Targets of Infection. Am. J. Pathol . 2003,163,2347-2370. [ C r ossRe f ] ) ( 12. Ryabchikova, E.I.; Kolesnikova, L.V.; Luchko, S.V. An Analysis of Features of Pathogenesis in Two Animal Models of Ebola Virus Infection. J. Infect. Dis. 1999,179, S199-S202.[C r o ssRef] ) ( 13. Martines, R.B.; Ng, D . L.; Greer, P.W.; Rollin, P . E.; Zaki,S.R. Tissue and cellular tropism, pathology andpathogenesis of E bola and Marburg viruses. J. Pathol. 2015,235,153-174. [ C r ossRef] [ Pu b Med ] ) ( 14. Messaoudi, I . ; Amarasinghe, G.K.; Basler, C.F. Filovirus pathogenesis and i m mune eva s ion: Ins i ghts fromEbola virus and Marburg virus. Nat. R ev. Microbiol. 2015,13,663-676. [ C rossRef] [ Pu b Med] ) ( 15. Falasca, L.; Agrati, C .;Petrosillo, N .;D i Caro, A. ; Capobianchi, M.R. ; I p polito, G.; Piacentini, M. Molec u lar mechanisms of Ebola vi r us pathogenesis: Fo c us on c ell death. Cel l Death Dif f er.2015, 22, 1250-1259. [ CrossRe f] [ Pu b Med ] ) ( 16 5 . . Zaki, S.R.; Goldsmith, C.S. Pathologic features of filovirus infections in humans. Curr. T op. Microbiol. Immunol. 1999,235,97-116. ) ( 17. Deen, G.F.; B routet, N.; X u, W.; Knust, B . ; Sesay, F.R.;McDonald, S.L.R.; Ervin, E.; Marrinan, J.E.; Gaillard, P.; Habib,N.; et al. Ebola RNA Persistence in Semen of Ebola Virus Disease Survivors 一 Final Report. N. Engl. J. Med.2017,377,1428-1437. [C rossRef] ) ( 18. Varkey, J .B.; Shantha, J .G.; Crozier, I. ; Kraft , C.S.; Lyon, G.M.; Mehta, A .K.; K umar, G. ; Sm i th, J.R.;Kainulainen, M.H.; Whitmer, S.; et al. Persistence of Ebola Virus in Ocular Fluid during Convalescence.N. Engl. J. Med. 2015, 372, 2423-2427. [ CrossRef ] ) ( 19. Sissoko, D .; K eita, M .; Diallo, B.; A liabadi, N . ; F i tter, D .L.; D ahl, B . A.; B o re, J.A.; K o undouno, F.R. ; Singethan, K.;Meisel,S.; et al. E bola virus persistence in breast milk after no reported il l ness: A likely source of virus transmission from mother to ch i ld. Clin. I n fect. D is. 2 017,64,513-516. [C r o ssRe f] ) ( 20. Vetter, P. ; Fischer, W.A. ; Schibler, M.; Jacobs, M. ; Bausch, D.G.; Kaiser, L . Ebola Virus Shedding andTransmission: Review of Current Evidence. J. Infect. D is. 2016,214, S177-S184. [C ro s sRef] ) ( 22. Liddell, A .M.; Davey,R.T.;Mehta, A.K.; Varkey, J.B.; Kraft, C.S.; Tseggay, G.K.; Badidi, O.; Faust, A.C.;Brown,K.V.; Suffredini, A .F.; et al.Characteristics and Clinical Management of a Cluster of 3 Patients WithEbola Virus Disease, Including the First Domestically Acquired Cases in the United States. Ann. Intern. Med. 2015, 163,81-90. [ CrossRef] ) ( 23. Nordenstedt, H.; Bah, E.I.; de la Vega, M.-A.; Barry, M.;N'Faly, M.; Barry, M.; Crahay, B.; Decroo, T.; VanHerp, M.;I n gelbeen, B. Ebola Virus in Breast Milk in an Ebola Virus-Positive Mother with Twin Babies,Guinea,2 0 15. Emerg. I n fect. Dis. 2016,22,759-760. [ CrossRe f ] ) ( 24. Brainard,J.; P ond, K.; Hooper,L.;Edmunds, K.; Hunter, P. Presence an d Persistence of Ebola or MarburgVirus i n Patients and S urvivors: A Rapid Systematic Review. PLoS Negl. Trop. Dis. 2 016, 10, e0004475. C rossRef l ) ( 25. MacIntyre, C.R.; Chughtai, A.A. R ecurrence and r e infection-a ne w paradigm for t he management of Ebolavirus d isease. Int. J. Infect. Dis. 2016 , 43,58-61. [ CrossRef] ) ( 26. Overholt, L.; Wohl, D.A.; Fischer, W.A.; Westreich, D.; Tozay, S. ; Reeves, E.; Pewu, K.; Adjasso,D.; Hoover, D.;Merenbloom,C.; et al. S t igma and Ebola survivorship in Liberia: Results from a longitudinal cohort stu d y.PLoS ONE 2018, 13, e0206595. [C rossR e f] ) ( 27. Chan,J.; Patel, M.; Tobin, S.; Sheppeard, V. Monitoring travellers from E b ola-affected countries in New SouthWales , A ustralia: What is the impact on travellers? BMC Public Health 2017,17,113. [Cr o s sRef] ) ( 28. Van Bortel, T.; Basnayake, A.; Wurie,F.; Jambai,M.;Koroma, A.S.;Mua n a, A.T.; Hann, K.;Eaton , J.;Martin, S.;Nellums, L.B. Psychosocial effects of an Ebola outbreak at individual, community and international levels.Bull. World Health Organ. 2016,94,210-214. [ C r o s sRef] ) ( 29. Davtyan, M.; B rown, B.;Folayan, M.O.Addressing Ebola-related Stigma: Le s sons Learned from HIV/AIDS.Glob. Health Action 2014,7,26058. [ C rossRef] ) ( 30. Mate, S.E.; Kugelman, J.R.; Nyenswah, T.G . ; Ladner, J.T.; Wiley, M. R .; Cordier-Lassalle, T.; Christie, A . ;Schroth, G.P.; Gross, S.M.;Davies-Wayne, G.J.; et al. Molecular Evidence of Sexual Transmission of EbolaVirus. N. Engl. J. Med. 2015,373,2448-2454. [C ro s sRef] ) ( 31. . . Christie,A.; D a vies-Wayne, G.J.; Cordier-Lassalle, T.; Cor d ier-Lasalle, T.; Blackley, D.J; Lane y , A.S.; Williams, D.E.; S hinde, S . A.; Badio, M.;Lo , T. ; et al. P ossible sexual transmission of Eb o la virus-Liberia,2015. MMWR Morb. Mortal. Wk l y. Re p . 2015,64, 479-481. ) ( 32. . Subissi, L.; K eita, M .; Mesfin, S . ; Rezza, G.;Di a llo, B. ; Van Gucht, S.; Mus a , E.O.; Yoti, Z.; K eit a , S.;Djingarey, M.H.; et al. Ebola Virus Transmission Caused by Persistently Infected Survivors of the 2014-2016Outbreak in West Africa. J. Infect. Dis. 2018,218, S287-S291. [Cr o s sRe f ] ) ( 33. Arias, A.; Watson, S.J.; Asogun, D.; Tobin, E.A.; Lu,J; Phan, M . V.T.; Jah, U.;Wadoum, R.E.G.;Meredith, L.;Thorne, L.; et a l . Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chainslinked to sporadic cases. V i rus Evol. 2 0 16, 2, vew016. [Cro ssRef] ) ( 34. Keita, M.; Duraffour, S.; Loman, N.J.; Rambaut, A.;Diallo, B.;Magassouba,N.; Carroll, M.W.; Quick,J.; Sall, A.A.; Glynn, J.R.; et al. Unusual Ebol a Virus Chain of Transmission, Conakry, Guinea, 2014-2015. E m erg.Infect . Dis. 2016,22,2149-2152. [ CrossRef ] [ P ubMed] ) ( 35. Diallo, B.; Sissoko, D.; Loman, N . J.; Bah, H.A.; B a h, H.; Worrell, M.C.; Conde, L.S.; Sacko, R.;Mesfin,S.;Loua, A.;et al. Resurgence of Ebola Virus Disease in Guinea Lin k ed to a Survivor With Virus Persistence inSeminal Fluid for More Than 500 Days. Clin. Infect. D i s. 2016,63,1353-1356 .[CrossRe f] [PubM e d] ) ( 36. Leroy, E.; Baize, S.; Volchkov, V.; Fisher-Hoch, S.; Georges-Courbot, M.-C . ; Lansoud-Soukate, J .; Capron, M.;Debre, P.; Georges, A.;McCormick, J. Human asymptomatic Ebola infe c tion and strong inflammatoryresponse. The Lancet 2000,355,2210-2215 . [Cr ossRef] ) ( 39. Dean, N.E.; H alloran, M.E.; Yang, Y.; L ongini, I.M. T ransmissibility and P a thogenicity of Ebola Virus: ASystematic Review and Meta-analysis of Household Secondary Attack Rate and Asymptomatic Infection.Clin. Infect. Dis. Off . Publ. Infec t . Di s . Soc. Am. 2016, 62, 1277-1286. [ CrossRef] [ P ubM ed ] ) ( 40. Richardson,E.T.; Kelly, J.D. ; Barrie, M.B.; Mesman, A.W.; Karku, S.; Quiwa, K.;Marsh, R.H.; Koedoyoma,S.; Daboh, F.; B arron, K.P.; et a l. Minimally Symptomatic Infection in an Ebola Hotspot': A Cross-Sectional Serosurvey. PLoS Negl. Trop. Dis . 2016,10,e0005087. [ CrossRef] ) ( 41. L 。 Clark,D.V.; Kibuuka,H.; Millard,M.; Wakabi,S.; Lukwago, L.; Taylor, A.; Elle r , M . A.;El l er, L.A.; Michael, N.L.;Honko, A.N.; et al. Long-term sequelae after Ebola virus disease in Bundibugyo,Uga n da: A retrospectivecohort study. Lancet Infect. Dis. 2015, 1 5,905-912. [ C r ossRef] ) ( 42. Rowe,A.K.; B e rtolli,J.; Khan, A.S. ; Mukunu,R.;Muyembe-Tamfum,J.J.; Bressler,D.; Williams,A.J.;Peters, C.J.;Rodriguez, L.; Feldmann, H.; et al. Clinical, Virologic, an d Immunologic Follow-Up of Convalescent EbolaHemorrhagic Fever Patients and Their Ho u sehold Contacts, Kikwit, Democratic Republic of the Congo. J. Infec t . Dis. 1999,179, S28-S35. [Cr o ssR e f ) ( 43. Bower, H.; G lynn, J . R. A systematic r e view a n d meta-analysis of se r oprevalence surveys of e bolavirusinfection. Sci. Data 2017,4,160133. [CrossRef ] ) ( 44. Houlihan, C.F.; M cGowan, C.R.; Dicks, S.; Baguelin, M.; Mo o re, D.A.J.;Mabey, D.;Rober t s, C.H.; Kumar, A.; Samuel, D.; T edder, R .; et al. Ebola e xposure, i llness experience, and E bola a ntibody prevalence i n international responders to the West African Ebola epidemic 2014-2016: A cross-sectional study. PLoS Med.2017, 14,e1002300 . [CrossRef ] [ Pub M ed] ) ( 45. Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200,373-383. [ C r oss R ef ] ) ( 46. Colombo,M.; Raposo, G.; Thery, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. R ev. C ell Dev. Biol. 2014, 30,255-289.[C rossRef] ) ( 47. Abels, E .R.; B reakefield, X.O. I ntroduction t o Extracellular Vesicles: Biogenesis, R N A Car g o Selection,Content, Release, and Uptake. C e ll Mol. N eurobiol. 2016, 36,301-312. [C rossRef] ) ( 48. Xu, R .; Greening, D.W.; Zhu, H. J ; Takahashi, N.; Sim p son, R.J. Extracellular ves i cle isolation an d characterization: Toward clinical application. J. Clin . Invest. 201 6 ,126,1152-1162. [ CrossRef] ) ( 49. Aker:s,J.C Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013,113,1-11.[ Cr o ssRef] ) ( 50. Keller, S.; Sanderson, M.P.; Stoeck, A.; Altevogt, P. Exosomes: From biogenesis and secretion to biologicalfunction. Immunol. Lett. 2006,107,102-108. [ Cros s Ref] ) ( l . S Schmidt,O.; Teis, D. The ESCRT ma c hinery. Cur r . Bio l . 2012, 22, R116-R120. [Cro ssRef] ) ( Henne, W.M.; B u chkovich, N.J; Emr, S. D . The ES C RT Pathway. Dev. Cell 2 011, 21,7 7 -91. [CrossR ef] 「 PubMed l ) ( 53. Stuffers, S.; Sem Wegner, C .; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence ofESCRTs. Traffic Cph. Den. 2009,10,925-937. [ Cro s sRef] ) ( 54. van N iel, G .; C harrin, S.; S imoes, S.; R omao, M.; Ro c hin, L. ; Saftig, P.; Marks, M.S.; R u binstein, E. ; Raposo, G. The Tetraspanin CD63 Regulates ESCRT-Independent and -Dependent Endosomal Sorting duringMelanogenesis. D e v. Cell 2011, 21,708-721. [ C r o s sRef] ) ( 55. Edgar, J.R.; Eden, E.R.; Futter, C . E. Hrs- and CD63-Dependent Competing Mechanisms Make Different SizedEndosomal Intraluminal Vesicles. Traffic 2014, 15, 197-211. [Cr oss R ef ] ) ( 56. Ghossoub, R.; Lembo, F . ;R u bio, A.; G a illard, C.B.;B o uchet, J ; Vi t ale, N . ; S l avik, J.; Machala, M.; Zimmermann, P.Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014,5,3477. [ C rossR ef ] ) ( 57. T rajkovic, K .; Hsu, C.; Chiantia, S.;Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons,M.Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319,1244-1247. [ CrossRef l ) ( 58. Alenquer, M.; Amorim,M.J. Exosome Biogenesis, Regulation, and F u nction in Viral Infection. Viruses 2015,7,5066-5083. [ C r o s s R e f ] ) ( 59. Vitelli, R.; Santillo, M . ; Lattero, D.;Chiariello, M.; Bifulco, M.; Bruni, C.B . ; Bucci, C. Role of the Small GTPaseRAB7 i n the Late Endocytic Pathway. J. Biol. C hem.1997,272,4391-4397. [Cros sR e f] ) ( 61. Baietti, M.F.; Zhang, Z.; M o rtier, E.;Me l chior, A.; Deg e est, G.; G eer a erts, A.; I v ars s on, Y.; De p oortere, F.;Coomans, C.; Vermeiren, E.; et al. S yndecan-syntenin-ALIX regu l ates the biog e nesis of exosomes. Nat. CellBiol. 2012, 14,677-685. [ Cr o ssRef] ) ( 62.2 . Ostrowski, M.; Carmo, N. B .; Kr u meich, S.; Fanget,I.; Raposo, G.; Sav i na, A.; Moi t a, C.F; Schauer, K.;Hume, A.N.; Freitas,R.P.; et al . Rab27a and Rab27b control different steps of the exosome secretio n pathway.Nat. C ell Biol. 2010, 12, 19-30.[ Cr o ssR e f] ) ( 63. Savina, A.;Vidal, M.; Colombo, M.I. The exosome pathway in K562 cells is regulated by Rab11. J. Cell Sci. 2002, 1 15,2505-2515. ) ( 64. Beckett, K.; Monier, S.; P almer, L.; Alexandre, C.;Green, H.; Bo n neil,E.; R aposo, G.; Thibault, P.; L e Bor g ne, R.;Vincent, J.-P. Drosophila S2 cells secrete wingless on exosome-like vesicles but the wingless gradient formsindependently of exosomes. Traffic Cph. Den. 2013, 14,82-96.[ CrossRef ] ) ( 65. Abrami, L.; Brandi, L.;Moayeri, M.; B rown, M.J.;K r antz, B.A.; Leppla, S.H.; van der Goot, F.G. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013,5,986-996. [ Cross R ef] ) ( 66. Koles, K.; Budnik, V. Exosomes go with the Wnt. Cell. Logist. 2012, 2, 169-173. [Cros s Ref] ) ( 67. H su, C.; M orohashi, Y.; Yoshimura, S.-I.; M anrique-Hoyos,N.; Jun g , S.; Lauterbach, M.A . ; Bakhti, M.; Gronborg, M.;Mobius, W.;Rhee,J.; et al. Regulation of exosome secretion by R ab35 and its GTPase-activatingproteins T BC1D1 0 A-C. J. Cell Biol. 2010, 189,223-232. [C r o ssRef] ) ( 68. Peinado, H .; .; Aleckovic, M.; Lavotshkin, S .; M atei, I .; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bonemarrow progenitor c e lls toward a pro-metastatic phenotype through M E T. Nat. Med. 20 1 2,18, 883-891. 「 CrossRe f ) ( 69. Bobrie, A.; Krumeich, S .; R eyal, F.; Recchi, C.; Moita, L.F.;Seabra, M.C.;Ostrowski, M.; Thery, C. Rab27asupports exosome-dependent and -i n dependent mechanisms that modify the t umor microenvironment andcan promote tumor progression. Ca n cer Res. 20 1 2, 72,4920-4930. [Cro ssRef] ) ( 70. Anderson, M.R.;Kashanchi,F;Jacobson, S. Exosomes in Viral Disease. Neurother. J. A m . Soc. Exp . Neurother.2016, 13,535-546.[C r o ssRef] ) ( 71. Schwab, A.; M eyering, S.S.; L epene, B.; Iordanskiy, S.; van Hoek, M.L.; Hakami, R . M.; K a shanchi, F.Extracellular vesicles from infected ce l ls: Potential for direct pathogenesis. Front. Microbiol. 2015, 6, 1132. CrossRe f l ) ( 72. Crenshaw, B . J.; Gu, L.;Sims, B.; Matthews, Q.L. Exosome Biogenesis and Biological Function in R e sponse toViral Infections. Open Virol. J. 2018,12,134-148. [ C r ossRef] ) ( 73. Votteler, J.; Sundquist, W.I. Virus Budding and the ESCRT Pathway. Cell Host Microbe2013, 1 4,232-241. C rossRef l ) ( 74. Ahmed, I.; Akram,Z.; Iqbal, H.M. N .;Munn, A.L. T he regulation of Endosomal Sorting Complex Required forTransport and accessory proteins in multivesicular body sorting and enveloped viral budding-An overview.Int. J. Biol. Macromol. 2019, 1 27,1 - 11. [ C r ossR e f] ) ( 75. Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. C e llular F u nctions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem. Sci. 2017,42,42-56. [Cross R e f] ) ( 76. Han, Z.;Madara,J.J;Liu, Y. ; L i u, W.;Ruthel, G.; Freedman, B.D.; Harty, R.N. ALIX Rescues Budding ofa Double PTAP/PPEY L-Domain Deletion Mutant of Ebola VP40: A R ole fo r ALIX in Ebola V i rus Egress.J. Infec t . Di s . 2015, 212 Suppl 2, S138-S145. [CrossRef ] ) ( 77. Licata, J .M.; Simpson-Holley, M.; Wright, N.T.;Han, Z.;Paragas,J;Harty, R.N. Overlapping Motifs (PTAP andPPEY) within the Ebola Virus VP40 Protein F unction Independently as Late Budding Domains: In v olvement of Host Proteins TSG101 a n d VPS-4. J. V i rol. 2003,77,1 8 12-1819.[Cro ssRef] ) ( 78. Martin-Serrano, J; Zang, T.; Bieniasz,P.D.HI V -1 and Eb o la virus encode sma l l pep t ide motifs that recruit T sg101 to sites of particle assembly t o f a cilitate egress. N at. M ed. 2001,7, 131 3 -1319. [Cr o s sRef] ) ( 79. Garrus, J .E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.;Stray, K.M.; Cote, M.;Rich, R.L.; et al. Tsg101 and the Vacuolar Protein Sorting Pathway Are Essential forHIV-1Budding. Cell 2001,107,55-65. [C r ossRef] ) ( 80. Ku, P .-I.;Bendjennat,M . ; Ballew, J;Landesman, M.B.; Saffarian, S. AL I X I s Recruited Tem p orarily into HIV-1Budding Sites at the End of Gag Assembly.PLoS ONE 2014, 9, e96950. [ C rossRef] ) ( 81. Carlton, J.G.; Agromayor, M.; Martin-Serrano, J. Di f ferential requirements for Alix and ESC R T-III incytokinesis and HIV-1 release. Proc . Natl.Acad. Sci. USA 2008, 105, 10541-10546 . [CrossRef ] ) ( 82. Strack, B.; Calistri, A.;Craig, S.;Popova, E.; Gottlinger, H.G. AIP1/ALIX Is a Binding Partner for HIV-1 p 6and EIAV p9 Functioning in Virus Budding. Cell 2003,114,689-699. [ C r ossRef] ) ( 83. Silvestri, L.S.; Ruthel, G.; Kallstrom, G.; Warfield, K.L.; Swenson, D.L. ; Nell e , T.; I v er s en, P.L.; Bava r i, S.; Aman, M.J. Involvement of Vacuolar Protein Sorting Pathway in Ebola Virus Release Independent of T SG101Interaction. J . Infect. Dis. 2007,196,S264-S270.[ Cross R ef ] ) ( 84. Goff, A.; Ehrlich, L.S.; Cohen, S.N.; Carter, C.A. Tsg101 Control of Human Immunodeficiency Virus T ype 1Gag Trafficking and Release. J. Viro l . 2003, 77,9173-9182. [Cro s sRe f ] [Pu b Med ] ) ( 85. Dussupt, V.; J avid, M.P.; Abou-Jaoude, G.; Jadwin, J.A.; Cruz, J. de L.; Nagashima, K.; B ouamr, F . TheNucleocapsid Region of HIV-1 Gag Cooperates with the PTAP and LYPXnL Late Domains to Recruit theCellular Machinery N ecessary for Viral Budding. PLoS Pathog. 2009,5, e1000339. [CrossRe f] [P ub Me d ] ) ( 86. Greening, D.W.; Xu, R.; Ji, H.;Tauro, B.J.;Simpson, R.J. A protocol for exosome isolation and characterization:Evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. MethodsMol. B iol. Clifton NJ 2015, 1295,179-209. ) ( 87. Konoshenko, M. Y .; Le k chnov, E.A.; Vlassov, A.V.; Laktionov, P.P. Is o lation of Extracellular Vesicles: GeneralMethodologies and Latest Trends. BioMed Res. I n t. 2018,2018,8545347.[C r o s s Ref ] ) ( 88. T aylor, D.D.; Shah, S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 2015,87,3-10. [ CrossRef ] ) ( 89. 3 Momen-Heravi, F; Balaj, L.; Alian, S.; Mantel, P.-Y.; Halleck, A.E.; Trachtenberg, A.J.; Soria, C.E.; Oquin, S.;Bonebreak, C.M.; Saracoglu, E.; et al. Current methods for the isolation of extracellular vesicles. Biol. Chem . 2013,394,1253-1262. [ C r ossRef] [ PubMed] ) ( 90. DeMarino, C.; Pleet, M.L.; Cowen, M.; Barclay,R.A.; Akpamagbo, Y.;Er i ckson, J.; Ndembe, N.; Charurat, M.;Jumare,J.; Bwala,S.; et al. Antiretroviral Drugs Alter the Content of Extracellular Vesicles from HIV-1-InfectedCells. Sci . Rep . 2018,8,7653.[ CrossRef] [ Pu b Med] ) ( 91. Timmins,J.;Scianimanico,S.; Schoehn, G.; Weissenhorn, W. Vesicular Release of Ebola Virus Matrix ProteinVP40. V i rology 2001, 283, 1-6. [C r o ssRef] ) ( 92. Kallstrom, G.; Warfield, K.L.; Swenson, D.L.; Mort, S.; Panchal, R.G.; Ruthel, G.; Bavari, S.; Aman, M.J. Analysis of Ebola virus and VLP release using an immunocapture assay. J. Virol. Meth o ds 2005, 127,1-9. [ C r ossRef l ) ( 93. Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.;Schmaljohn, A.; Aman, M.J. L i pid R a ft Microdomains: A Gateway for Compartmentalized Trafficking ofEbola and Marburg Viruses. J. Exp. Med. 2 002,1 9 5,593-602. [Cr ossRef] ) ( 94. Beniac, D.R.; Melito, P.L.; deVarennes,S.L.; Hiebert, S.L.; Rabb, M.J.;Lamboo, L.L.; Jones, S.M.; Booth, T.F.The Organisation of Ebola Virus Reveals a Capacity fo r Extensive, Modular Polyploidy. PLoS ONE 2012, 7, e29608.[ CrossRef ) ( 95. Pleet, M.L.; Mathiesen, A.; DeMarino,C.; Akpamagbo, Y.A.; Barc l ay, R.A.; Schwab, A.; Iordanskiy, S.;Sampey, G.C.; Lepene, B.; Nekhai, S.; et a l . Ebola VP40 in Exosomes Can Cause Immune Cell Dysfunction.Front. Microbiol. 2016, 7 ,1765. [ C r o s sRef] ) ( 96. Pleet, M.L.; Erickson, J.;DeMarino, C.; Barclay, R . A.; Cowen, M.; Lepene, B.; Lia n g,J; Kuhn , J.H.;Prugar,L.I.;Stonier, S.W.; et al. Ebola Virus VP40 Modulates Cell Cycle and Biogenesis of Extracellular Veiscles. J InfectDis. 2018,218, S365-S387. [ C rossR e f] ) ( 97. . Cantin, R.; Diou, J.;Belanger, D.; Tremblay, A.M.; Gilbert, C. Discrimination between exo s omes and HIV-1:Purification of both vesicles from cell-free supernatants. J. Immunol. Methods 2008 , 338, 21-3 0 . [Cross Ref] ) ( 98. Pleet, M.L.;DeMarino, C.; Lepene, B . ; Aman, M.J.; Kashanchi, F. The Role of E x osomal VP 4 0 in E bola VirusDisease. DNA Cell Biol. 2017,36,243-248. [ Cro s sRef ] ) ( 99. Timmins, J.;Schoehn, G.; R i card-Blum, S.; Scianimanico, S.; Vernet, T.; Ruigrok, R.W.H.; W e issenhorn, W. Ebola Virus Matrix P rotein VP40 Interaction with Human Cellular Factors Tsg101 and N e dd4. J. Mol. B i ol.2003,326,493-502. [C rossRef] ) ( 100. H artlieb, B.; Weissenhorn, W. Filovirus assembly and budding. Virology 2006,344,64-70.[C r o ssRef] ) ( 101. Jasenosky, L.D.; Neumann, G.; Lukashevich,I.; Kawaoka, Y. Ebola Virus VP40-Induced Particle Formation and Association with the Lipid Bilayer. J. Virol. 2001,75,5205-5214. [Cro ssRef] ) 104. Noda,T.; Ebihara, H.; Muramoto, Y.; Fujii, K.; Takada, A.; Sagara,H.; Kim, J.H.; Kida, H.; Feldmann, H.;Kawaoka, Y. Assembly and Budding of Ebolavirus. PLoS Pathog. 2006,2, e99. [CrossRef] 105. Surjit, M.; Oberoi, R.; Kumar,R.; Lal, S.K. Enhanced α1 Microglobulin Secretion from Hepatitis E VirusORF3-expressing Human Hepatoma Cells Is Mediated by the Tumor Susceptibility Gene 101. J. Biol. Chem.2006,281,8135-8142. [CrossRef] ( 106. N agashima, S.; T a kahashi,M.; Jirintai, S.; Tanaka, T.;Nishizawa, T. ; Y a suda, J; Okamoto, H. T um o ur susceptibility gene 101 and the vacuolar protein sorting pathway are required for the release of hepatitis E virions. J. Gen. Virol. 2011, 92,2838-2848.[Cr os sRe f ] ) ( 107. P ark, A.; Yun, T.;Vigant, F.;Pernet, O.; Won, S.T.; Dawes, B.E.; B a rtkowski, W.; Freiberg, A.N.; Lee, B. Ni p ahVirus C P rotein Recruits Tsg101 to Promote the Efficient Release of Virus in an ESCRT-Dependent Pathway.PLoS P athog. 2016,12,e1005659.[C rossRef ] ) ( 108 . Bissig, C.; Gruenberg, J. ALIX and the multivesicular endosome: ALIX in Wonderland. Tre n ds Cell Biol. 2 0 14, 24,19-25. [ Cr o ssRef] ) ( 109. Zhai, Q.;Fisher, R .D.; Chung, H.-Y.; Myszka, D.G.; Sundquist, W.I.; H i ll, C.P. Structural and functional studies of ALIX interactions with YPXnL late domains of HIV-1 and EIAV. Nat. Struct. Mol. Biol.2008, 15, 43-49. [ CrossRef ) ( 110. Yasuda,J;Nakao,M.; Kawaoka, Y.; Shida, H. Nedd4 Regulates Egress of Ebola Virus-Lik e Particles fromHost Cells. J. Virol. 2003,77,9987-9992. [ CrossRef ] ) ( 111. M alakhova,O.A.;Zhang,D.-E. ISG15 Inhibits Nedd4 Ubiquitin E3 Activity and Enhances the Innate AntiviralResponse. J. Biol. Chem. 2008,283,8783-8787. [Cr ossRef] ) ( 112. H an, Z.;Sagum, C.A.; Bedford, M. T .; S idhu, S.S.; Sudol, M.; H arty, R.N. I TCH E 3 Ubiquitin Ligase Interactswith Ebola Virus VP40 To Regulate Budding. J . V irol. 2016,90,9163-9171. [ C rossRef] ) ( 113. Han,Z.;Sagum,C.A.;Takizawa,F.;Ruthel, G.; Berry,C.T. ; Kong,J.;Sunyer,J.O.; Freedman, B.D.; Bedford, M.T.;Sidhu, S.S.; et al. Ubiquitin Ligase WWP1 Interacts with Ebola Virus VP40 To Regulate Egress. J. Virol. 2017, 91,e00812-17. [ CrossRef] ) ( 114. Okumura, A.;Rasmussen, A.L.; Halfmann, P.;Feldmann, F; Yoshimura, A.; Feldmann, H.;Kaw a oka, Y.;Harty, R .N.; Katze, M.G. Suppressor of Cytokine Signaling 3 Is an Inducible Host Factor That Regulates Virus Egress during Ebola Virus Infection. J . Virol . 2015,89,10399-10406.[Cr o s sRef] ) ( 115. N ascimento,R.; Costa,H.; Parkhouse, R.M.E. Virus manipulation of ce l l cycle. Pro t oplasma 2012,249,519-528. 「 CrossRe f l ) ( 116. F ehr, A.R.; Yu, D. Control the Host Cell Cycle: Viral Regulation of the Anaphase-Promoting Complex. J. Virol. 2013,87,8818-8825. [ CrossRef ] ) ( 118. C haurushiya, M.S.; Weitzman, M.D. Viral manipulation of D NA repair and cell cycle checkpoints. DNA Repair 2009, 8,1166-1176. [ C rossRef] ) ( 119. S antiago, F; Clark, E.; Chong, S.; Molina, C.; Mozafari, F.; Mahieux, R.; Fujii, M.; Azimi, N.; Kashanchi,F. T ranscriptional Up-Regulation o f the Cyclin D2 Gene and Acquisition of New Cyclin-Dependent Kinase Partners in Human T-Cell Leukemia Virus Type 1-Infected Cells. J.V i r o l. 19 9 9, 73,9917-9927. ) ( 120. K ehn, K.;Deng, L.; de la Fuente, C.; Strouss, K.; Wu, K.;Mad d ukuri, A.; Baylor, S.; Rufner, R.; Pumfery, A.;Bottazzi, M.E.; et al. The role of cyclin D2 and p21/waf1 in human T-cell leukemia virus type 1 infected cells. Retrovirology 2004, 1, 6. [ CrossRef] ) ( 121. Zhou, L.;Kang, D.;Xu, C.; Zhao, W.; Tian, B.; Chen, L. Expression of cyclin D1 and cyclin E significantlyassociates with human papillomavirus subtypes in Bowenoid papulosis. Acta Histochem. 2013, 115, 339-343. 「 CrossRe f l ) ( 122. R ipple, M.J.; Parker Struckhoff, A.; Trillo-Tinoco,J; Li, L.;Margolin, D.A.; McGoey, R.; Valle, L.D. Activationof c-Myc and Cyclin D 1 by JCV T-Antigen and B- C atenin in Colon Cancer. PL o S ONE 2014 , 9, e10 6 257. [ CrossRe f ) 124. Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus encoded LMP1 regulatescyclin D1 promoter activity by nuclear EGFR and STAT3 in CNE1 cells. J. Exp. Clin. Cancer Res. CR 2013, 32,90.[CrossRef] ( 125. C asimiro,M.C.; Velasco-Velazquez,M.; Agu i rre-Alvarado, C.; P estell, R.G.Overview of cyclins D1 functionin cancer and the CDK inhibitor landscape: Past and present. E x pert Opin. I n vestig. Drugs 2014,23,295-304. [ CrossRef l ) 126. Pestell,R.G. New Roles of Cyclin D1. Am. J. Pathol. 2013,183,3-9. [CrossRef] ( 127. Klein,E.A.; Assoian, R.K. Transcriptional regu l ation of th e cyclin D1 gene a t a glance. J. Cell Sci. 2008,121,3853-3857. [ C rossRef ] ) ( 128. B B jorndal, A.S.; Szekely, L.; Elgh,F. Ebola v irus infection inversely correlates with the overall expressionlevels of promyelocytic leukaemia (PML) pr o tein in cultured cells. BMC Microbiol. 2003,3, 6. [Cro s s Ref] ) ( 129. Nanbo, A.; Watanabe,S.; Halfmann, P.; Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and R NA in infected cells. S ci. Rep. 2013,3, srep01206. [ CrossR e f ] ) ( 130. V ecchio,K.D.; Frick, C.T.; Gc, J.B.; Oda, S.;Gerstman, B.S.;Saphire,E.O.;Chapagain, P.P.; Stahelin, R.V. A cationic, C-terminal patch and s t ructural r e arrangements in Ebola virus mat r ix VP40 protein control its interactions with phosphatidylserine. J. Biol . Chem. 2018, 293, 3335-3349. [Cross R ef ] ) ( 131. K ota,K.P.; B enko, J.G.;Mudhasani, R.; Retterer, C.; Tran, J.P.; Bavari, S.;Panchal, R.G. High Content ImageBased Analysis Identifies C e ll Cycle Inhibitors as Regulators of Ebola Vir u s Infection. Viruses 2012, 4, 1865-1877. [ CrossR e f ] ) ( 132. W auquier, N.;Becquart, P.; Padilla,C.; Ba i ze, S.; Leroy, E.M. H u man Fata l Zaire Ebola Virus Infection Is Associated with an Aberrant Innate Immunity and with Massive Lymphocyte Apoptosis. PL o S Negl. Trop.Dis. 2010,4, pii: e837.[ CrossRef] ) ( 133. Gupta, M.; Spiropoulou, C.; Rollin, P.E. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Vir o logy 2007, 364, 45-54.[Cros sRef] ) ( 134.Y Yaddanapudi, K.; Palacios, G.;Towner, J.S.; Chen, I.; Sariol, C.A.;Nichol, S.T.; Lipkin, W.I. Impli c ation of a retrovirus-like glycoprotein peptide in t he immunopathogenesis of Ebola and Marburg viruses. FASEB J . 2006,20,2519-2530. [C rossRef] ) ( 135. M artin, B .; Reynard, O.; Vo l chkov, V.; De c roly, E. F il o virus proteins for antiviral dru g discovery: Structure/function o f p roteins involved i n assembly an d bud d ing. Antiv i ral Res. 2018, 150,183-192. CrossRe f l ) ( 136. W atanabe, S.; Noda, T.; Kawaoka, Y. Functional Ma p ping of the Nucleoprotein of Ebola Virus. J. Virol. 2006,80,3743-3751.[C rossRef ] ) ( 137. Huang, Y.;Xu, L.;Sun, Y.; Nabel,G.J. The Assembly of Ebola Virus Nucleocapsid Requires V irion-AssociatedProteins 35 and 24 and P osttranslational Modification of Nucleoprotein. Mol. Cell 2002, 1 0,307-316. C rossRef l ) ( 138. K irchdoerfer,R.N.; Abelson,D.M.; Li, S.; Wood,M.R.; Saphire,E.O. Assembly of the Ebola Virus Nucleoproteinfrom a Chaperoned VP35 Complex. Cell Rep. 2015, 12,140-149.[ CrossRef ] ) ( 139. B harat, T.A.M.;Noda, T.;Riches, J.D.; Kraehling, V.;Kolesnikova, L.;Becker, S.; Kawaoka, Y.; Briggs, J.A.G. Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography. Proc. Natl. Acad. Sci . USA 2012,109, 4275-4280.[C r o ssR e f] ) ( 140. M artin, B.; Canard, B . ; Decroly, E. F ilovirus proteins for antiviral drug discovery: Structure/function bases of the replication cycl e . Antiviral Res.2017,141,48-6 1 . [CrossRe f ) ( 141.I Dong, S . ; Yang, P; Li, G.; Liu, B.; Wang, W.;Liu,X.;Xia, B.; Yang, C.; Lou, Z.; Guo, Y.; et al. Insight into theEbola virus nucleocapsid assembly mechanism: Crystal structure of Ebola virus nucleoprotein core do m ain at 1.8 A resolution. Protein Cell 2015,6,351-362. [ C r o s sRef ) ( 143. Noda, T.; Hagiwara, K.; Sagara,H.; Kawaoka, Y.Characterization of the Ebola virus nucleoprotein-RNAcomplex.J. G en. Virol. 2010,91 , 1478-1483.[ CrossRef] ) ( 144. Guo, Y.; W ang, W.; Ji, W.; D e ng, M.; Su n , Y.; Zhou, H.; Yan g , C.; Den g , F.; Wang, H.; Hu, Z . ; et al . Crimean-Congo hem o rrhagic fever viru s nucl e oprotein reveals endonuclease activity in bunyaviruses. Proc. Natl. Acad. Sci . USA 2012,109,5046-5051. [Cr ossRef] ) ( 145. H astie, K.M.;Kimberlin, C.R.; Zandonatti,M.A.;Mac R ae, I.J.; Saphire, E.O. Structure of the Lassa virusnucleoprotein reveals a dsRNA-specific 3' to 5'exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108,2396-2401.[C ros s Ref] ) ( 146. Qi, X.;Lan, S.;Wang, W.; Schelde, L.M.; D ong, H.; Wallat, G.D.; Ly, H.; Liang, Y.;Dong, C. Cap binding andimmune evasion revealed by Lassa nucleoprotein structure. Nature 2010,468,779-783. [C r o s s Ref] ) ( 147. H atakeyama,D.;Ohmi, N.; Saitoh, A.; Makiyama, K.; Mo r ioka, M.;Okazaki,H.;Kuzuhara, T. Acetylation oflysine residues in the recombinant nucleoprotein and VP40 m a trix p r otein of Zaire E bolavirus by eukaryotichistone acetyltransferases. B iochem. Biophys. Res. Commun. 2018, 504,635-640. [Cro s s R e f] ) 148.FFujisawa,T.;Filippakopoulos,P. Functions ofbromodomain-containing proteins and their roles in homeostasisand cancer. Nat. Rev. Mol. Cell Biol. 2017,18,246-262. [CrossRef ( 149. Ren, W.; W a ng, C.;Wang, Q.; Zhao, D.; Zhao, K.;Su n , D.; Li u , X.;Han, C . ; Hou, J.; Li, X.; et al. Bromodomainprotein Brd3 promotes Ifnb1 transcription v i a enhancing IRF3/p300 complex formation and recru i tment toIfnb1 promoter in macrophages. Sci. Rep. 2017,7, 39986. [Cr o s s Ref] ) ( 150. G arcia-Dorival,I . ; Wu, W.; Armstrong,S.D.; B a rr, J.N.; Carroll, M.W.; Hewson, R .; Hiscox, J .A. Elucidation of the Cellular Interactome of Ebola Virus Nucleoprotein and Identification of Therapeutic Targets. J. Proteome Res. 2016, 15,4290-4303. [C rossRef] ) ( 151. P roserpio,V.; Fittipaldi,R.; Ryall, J.G.; Sartorelli, V.; Caretti, G. The methyltransferase SMYD3 m ediatesthe recruitment of transcriptional cofactors at the myostati n and c-Met genes and regulates skeletal muscleatrophy. Genes Dev. 2013,27,1299-1312. [ CrossRef] ) ( 152. Ebihara,H.;Takada, A.;Kobasa,D.;Jones,S.; Neumann, G.; Theriault, S.; Bray, M.; Feldmann,H.;Kawaoka, Y.Molecular Determinants of Ebola Virus Virulence in Mice. PLo S Pathog. 2006, 2, e73.[Cro s s R e f] ) 153. Hoenen,T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.;Feldmann,H. Inclusion Bodies Are a Site of Ebolavirus Replication. J. Virol. 2012,86,11779-11788. [CrossRef] 154. Martin, B.; Hoenen, T.; Canard, B.; Decroly, E. Filovirus proteins for antiviral drug discovery: Astructure/function analysis of surface glycoproteins and virus entry. Antiviral Res. 2016,135,1-14.[CrossRef] 155. Miller, E.H.; Chandran, K. Filovirus entry into cells - new insights. Curr. Opin. Virol. 2012,2, 206-214.[CrossRef| 156. Simmons, G. Filovirus Entry. In Viral Entry into Host Cells; Advances in Experimental Medicine and Biology;Springer: New York, NY, USA, 2013; pp.83-94. ISBN 978-1-4614-7650-4. 157. Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular Entry of Ebola Virus Involves Uptake by aMacropinocytosis-Like Mechanism and Subsequent Trafficking through Early and Late Endosomes. PLoSPathog. 2010,6,e1001110.[CrossRef] 158. Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y.Ebolavirus Is Internalized into Host Cells via Macropinocytosis in a Viral Glycoprotein-Dependent Manner.PLoS Pathog. 2010,6,e1001121. [CrossRef] ( 159. W hite, J.M.; Schornberg, K.L. A new player in the p u zzle of fil o virus entry. Nat. Rev. M i crob i ol. 2012,10, 317-322. [ Cros s Re f ) 160. Markosyan, R.M.; Miao, C.; Zheng, Y.-M.; Melikyan, G.B.; Liu, S.-L.; Cohen, F.S. Induction of Cell-Cell Fusionby Ebola Virus Glycoprotein: Low pH Is Not a Trigger. PLoS Pathog. 2016, 12,e1005373. [CrossRef] 161..VVolchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.-D. Processing of the Ebola virus glycoprotein bythe proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762-5767. [CrossRef] ( 162. J effers, S.A.; Sanders, D.A.; Sanchez, A. Covalent Modifications of the Ebola Virus Glycoprotein.J. Viro l . 2002,76,12463-12472. [C rossR e f] ) 163. Gruenberg,J.;Stenmark, H. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 2004,5,317-323. [CrossRef] ( 164. G adila, S.K.G.; Kim, K. Cargo t rafficking from the trans-Golgi network towards the endosome. Biol. C e ll2016,108,205-218.[C rossRef] ) 167. Escudero-Perez, B.; Volchkova, V.A.; Dolnik, O.; Lawrence, P.;Volchkov, V.E. Shed GP of Ebola Virus TriggersImmune Activation and Increased Vascular Permeability. PLoS Pathog.2014,10,e1004509. [CrossRef] 168.ILai, C.-Y.; Strange, D.P.; Wong, T.A.S.; Lehrer, A.T.; Verma, S. Ebola Virus Glycoprotein Induces an InnateImmune Response In vivo via TLR4. Front. Microbiol. 2017,8,1571.[CrossRef] 169. Okumura, A.; Pitha,P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola Virus Glycoprotein andHost Toll-Like Receptor 4 Leads to Induction of Proinflammatory Cytokines and SOCS1. J. Virol. 2010,84,27-33. [CrossRef] 170. Iampietro,M.; Santos,R.I.; Lubaki,N.M.; Bukreyev, A. Ebola Virus Shed Glycoprotein Triggers Differentiation,Infection, and Death of Monocytes Through Toll-Like Receptor 4 Activation. J. Infect. Dis. 2018,218, S327-S334.TCrossRefl ( 171. M artinez, O.; J ohnson, J.C.; Honko, A.; Yen, B.;Shabman, R.S.; Hensley, L.E.; Olinger, G.G.; B a sler, C .F.Ebola Virus Exploits a Monocyte Differentiation Program To Promote Its Entry.J. Virol. 2013,87, 3801-3814. 「 CrossRe f l ) ( 172. Edri, A.; Shemesh, A.; Iraqi, M.;Matalon, O.; Brusilovsky, M.; Hadad, U.;Radinsky, O.;Gershoni-Yahalom, O.; Dye,J.M.;Mandelboim, O.; et al. The Ebola-Glycoprotein Mo d ulates the Function of Natural Killer Cells. Front. Immunol. 2018,9,1428. [ C rossRef] ) ( 173. I ampietro, M.; Younan, P. ; N i shida, A.; Dutta, M.; Lu b aki, N.M.; Santos, R.I.; Koup, R.A.; Katze, M.G.;Bukreyev, A. Ebola v i rus glycoprotein directly triggers T lymphocyte death despite of the lack of infection.PLoS Pathog.2 0 17, 1 3 , e1006397. [C rossRef] ) ( 174. Maruyama,T.; Par r en, P.W.H.I.; Sanchez, A.;Rensink, I.;Rodriguez, L.L.;Khan, A.S.;Peters,C.J.; Burton, D.R.Recombinant Human Monoclonal Antibodies to Ebola Virus. J. Infect. Dis. 19 9 9, 179,S235-S239. [Cr o s sRef] ) ( 175. Mohan, G.S.; Li,W.; Ye, L.; Compans,R.W.; Yang, C. Antigenic Subversion: A Novel Mechanism of HostImmune Evasion by Ebola Virus. PLoS Pathog. 2012,8, e1003065. [Cro s s Ref] ) ( 176. B E ixler, S.L.; G off, A. J . The Role of Cytokines and Chemokines i n Filovirus Infection. V iruses 2015, 7,5489-5507. CrossRe f ) ( 177. V illinger, F.; Rollin, P.E. ; Brar, S.S . ; Chikkala , N.F.; Winter,J.; Sundstrom, J.B.; Zaki, S.R.; Swanepoel, R . ; Ansari, A.A.; Peters, C.J. Markedly Elevated Levels of Interferon (IFN)-Y, IFN-α, Interleukin (IL)-2, IL-10, and Tumor Necrosis Factor-a Associated with Fatal Ebola Virus Infection. J. Infect. Dis. 1999 , 179, S188-S191. 「 CrossRef l ) ( 178. BEaize, S.; Leroy, E.M.; Georges, A.J.; Georges-Courbot, M.C.; Capron, M.; Bedjabaga, I;Lansoud-Soukate,J.;Mavoungou, E. In f lammatory responses in Ebola virus-infected patients. Cli n . Exp. Imm u nol. 2002 , 128,163-168. [ CrossRef ] ) ( 179. T rinchieri, G. Interleukin-12 and i nterferon-gamma. Do they always go together? Am. J . Pa t hol. 1995,147, 1534-1538. ) 180. Szabo, S.J.; Dighe, A.S.; Gubler, U.; Murphy, K.M. Regulation of the Interleukin (IL)-12R B2 SubunitExpression in Developing T Helper 1 (Th1) and Th2 Cells. J. Exp. Med. 1997,185,817-824. [CrossRef] ( 181 . Otani,T.;Nakamura, S.; Toki, M.;Motoda,R.; K urimoto,M.; Orita, K. Identification of IFN-gamma-producingcells in IL-12/IL-18-treate d mice . Cel l . Immunol . 1999,198,11 1 -119.[C ro s sRef] ) ( 182. W ei, Y.P.; Kita, M.; Shinmura, K.; Yan, X.Q.;Fukuyama, R.;Fushiki, S.; Imanishi, J. Ex p ression of IFN-gammain cerebrovascular endothelial cells fro m aged mice. J. Interferon Cytokine Res. Off. J. Int. Soc. Interfe r onCytokine R es. 2000, 20, 403-409. [ CrossR e f] ) ( 183. S uo, Z.; Tan, J.;Placzek, A.; Crawford,F.;Fang, C.;Mullan, M. Alzheimer’s beta-amyloid peptides induceinflammatory cascade in human vascular cells: Th e roles of cytokines and CD40. Brai n Res. 1998 , 807, 1 1 0-117. [ CrossRef ] ) ( 184. Hakonarson, H.; Maskeri,N.; Carter, C.; Grunstein, M .M. R e gulation of T H 1- and TH2-type cyt o kine expression a nd action in atopic a sthmatic sensitized a irway smooth mu s cle. J. C l i n. Invest. 1 9 9 9, 103, 1077-1087. [ CrossRef] ) ( 186.B Baize, S.; Leroy, E.M.; Georges-Courbot, M.-C.; Capron, M.; Lansoud-Soukate, J.;Debre, P.; F isher-Hoch, S.P.;McCormick, J.B.; G eorges, A .J. Defective humoral responses and extensive intravascular apoptosis areassociated with fatal outcome in E bola virus-infected patients. Nat. Med. 1 999,5,423-426.[Cro s sRef ) ( 187. S teel,J.C.; Waldmann, T.A.;Morris, J.C. Interleukin-15 biology and its the r apeutic implications in cancer. Trends Pharmacol. Sci. 2012, 33,35-41. [CrossRef ) 188. Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1(MCP-1): AnOverview. J. Interferon Cytokine Res. 2009,29,313-326.[CrossRef] 189. Kindrachuk,J.; Wahl-Jensen, V.; Safronetz, D.; Trost, B.; Hoenen, T.;Arsenault, R.; Feldmann, F.;Traynor, D.;Postnikova, E.; Kusalik, A.; et al. Ebola Virus Modulates Transforming Growth Factor B Signaling andCellular Markers of Mesenchyme-Like Transition in Hepatocytes. J. Virol. 2014,88,9877-9892.[CrossRef] ( 192. Z hang, B.; Shen, L.; Shi, H.; Pan, Z.; Wu, L.; Yan, Y.; Zhang, X.; Mao, F.;Qian, H.; Xu, W. Exosomes fromHuman Umbilical Cor d Mesenchymal Stem Cells: Identification, Purification, and Biological Characteristics. Stem Cells Int. 2016,2016,1929536. [C rossRef] ) ( 193. Nickel, W.; Rabouille, C. Mechanisms of regulated unconventional protein secretion. Nat. Re v . Mol. Cell Bio l .2009, 1 0,148-155.[C rossRef] ) ( 194. K onadu, K.A.; Chu, J.; Huang, M.B.; Amancha,P.K.; Armstrong, W.; Powell, M.D.; Villinger, F.; Bond, V.C.Association of Cytokines With Exosomes in the Plasma of HIV-1-Seropositive Individuals. J. Infect. D i s. 2015,211,1712-1716. [ CrossRef] ) ( 195. F itzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of CytokinesEncapsulated in ExtraCellular Vesicles. S c i. Rep. 2018,8, 8973. [ C r ossRe f ] ) ( 196. S S mith, J.R.; Todd, S.; Ashander, L.M.; Charitou, T.; Ma, Y.; Yeh, S.; Crozier, I . ; M ichael, M . Z.; Appukuttan, B.;Williams, K.A.;et al. Retinal Pigment Epithelial C ells are a Potential Reservoir for Ebola Virus in the HumanEye. Transl. Vis. S ci. T echnol. 2 017, 6, 12. [C r o ssR e f] ) ( 197. Z eng, X.; Blancett, C.D.; Koistinen, K.A.; Schellhase, C. W .; Bearss, J.J.; R a doshitzky, S.R.; Ho n nold, S.P.; Chance, T.B.; Warren, T.K.; F roude, J.W.;et al. Identification and pathological characterization of persistent asymptomatic Ebola virus infection in rhesus monkeys. Nat. M icrobiol. 2017,2,17113. [ CrossRef] ) @ 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open accessCC article distributed under the terms and conditions of the Creative Commons Attribution(CC BY) license (http://creativecommons.org/licenses/by/4.0/). 在急性感染过程中,埃博拉病毒(EBOV)会引发从无症状表现到急性出血热等多种症状。而且,幸存者在无症状的恢复期也可能会传播病毒。在急性感染中,可能会发生细胞因子风暴(Cytokine Storm)和淋巴细胞凋亡,导致被感染者出现不可控的系统性炎症。近的研究证实,在感染过程中,埃博拉病毒蛋白VP40、糖蛋白(GP)和核蛋白(NP)会被装载到细胞外囊泡中。含有埃博拉病毒蛋白的细胞外囊泡已被证实能诱导受体免疫细胞凋亡,并且含有促炎性细胞因子。本篇文章中,研究者梳理了当前与埃博拉病毒相关的细胞外囊泡的研究现状,包括细胞外囊泡的生物发生机制、内含物以及它们对受体细胞的影响。另外,研究者讨论了埃博拉病毒所感染细胞产生的细胞外囊泡可能会引发的一些影响,提出了有待解决的问题和未来的研究方向。
确定










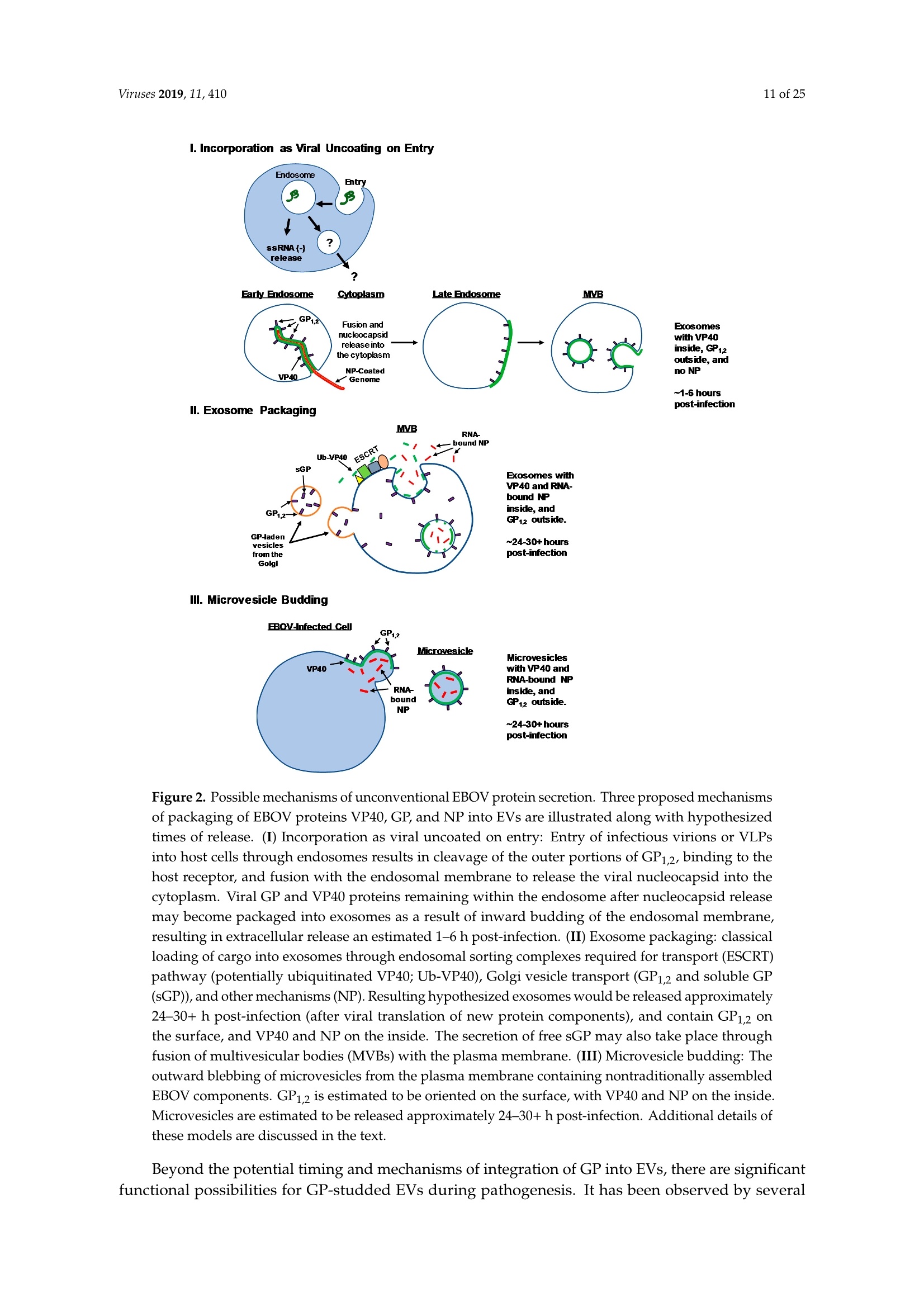














还剩23页未读,是否继续阅读?

产品配置单

大昌华嘉科学仪器为您提供《细胞外囊泡和埃博拉病毒中一种新的免疫逃避机制检测方案(Zeta电位仪)》,该方案主要用于其他中一种新的免疫逃避机制检测,参考标准--,《细胞外囊泡和埃博拉病毒中一种新的免疫逃避机制检测方案(Zeta电位仪)》用到的仪器有Microtrac纳米粒度及Zeta电位分析仪
推荐专场






相关方案
更多
该厂商其他方案
更多