









Agilent 1200系列纯化系统是药物高通量合成小分子和大分子化合物库纯化的通用工具,能够得到高纯度馏分,获得目标化合物的高回收率。Agilent 6110/6120系列MSD是新一代单级四极杆仪器,可以进行简便而可靠的质谱引导的馏分收集,也可以完成更复杂的纯化工作,如组合离子源或使用正/负极切换等。
方案详情

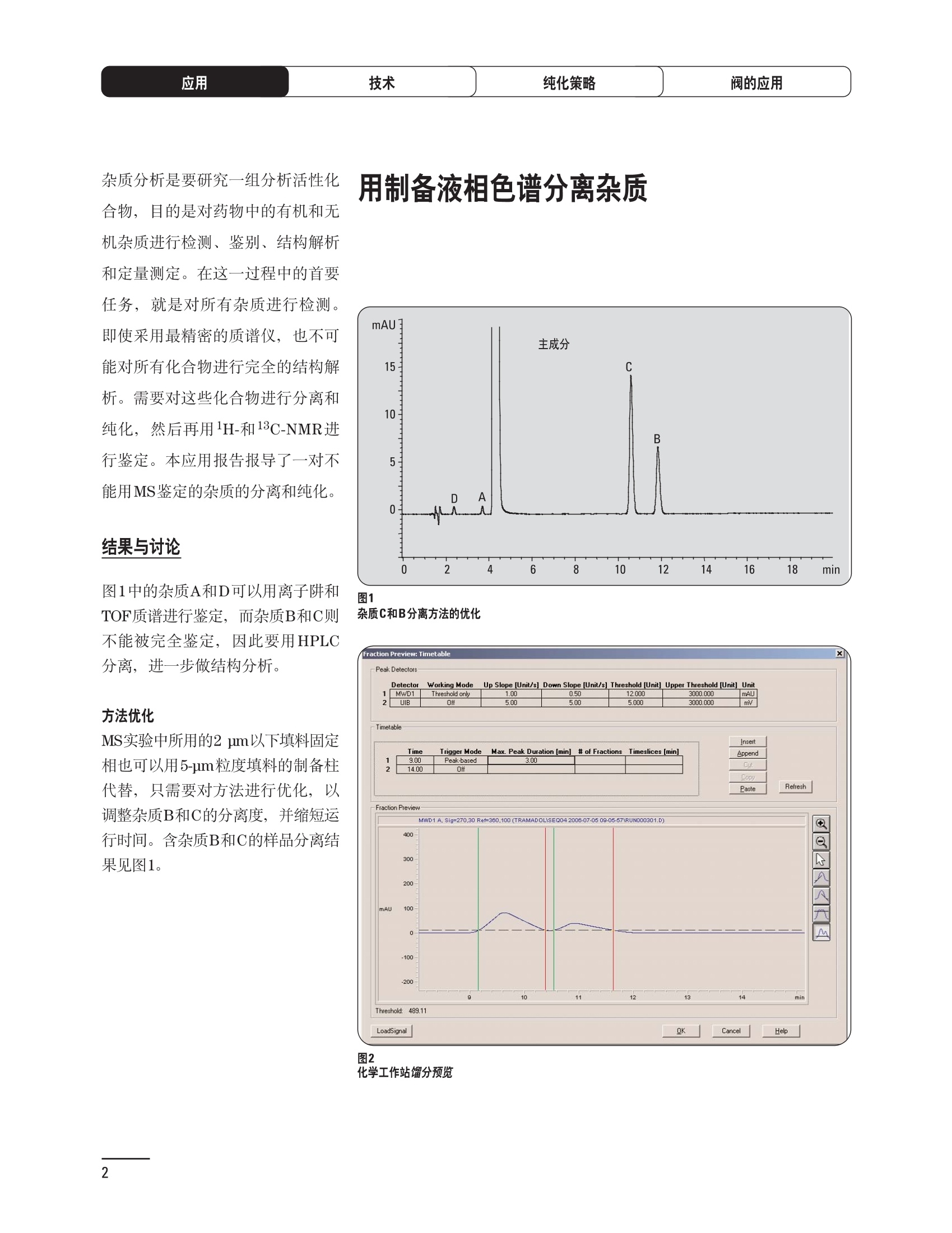
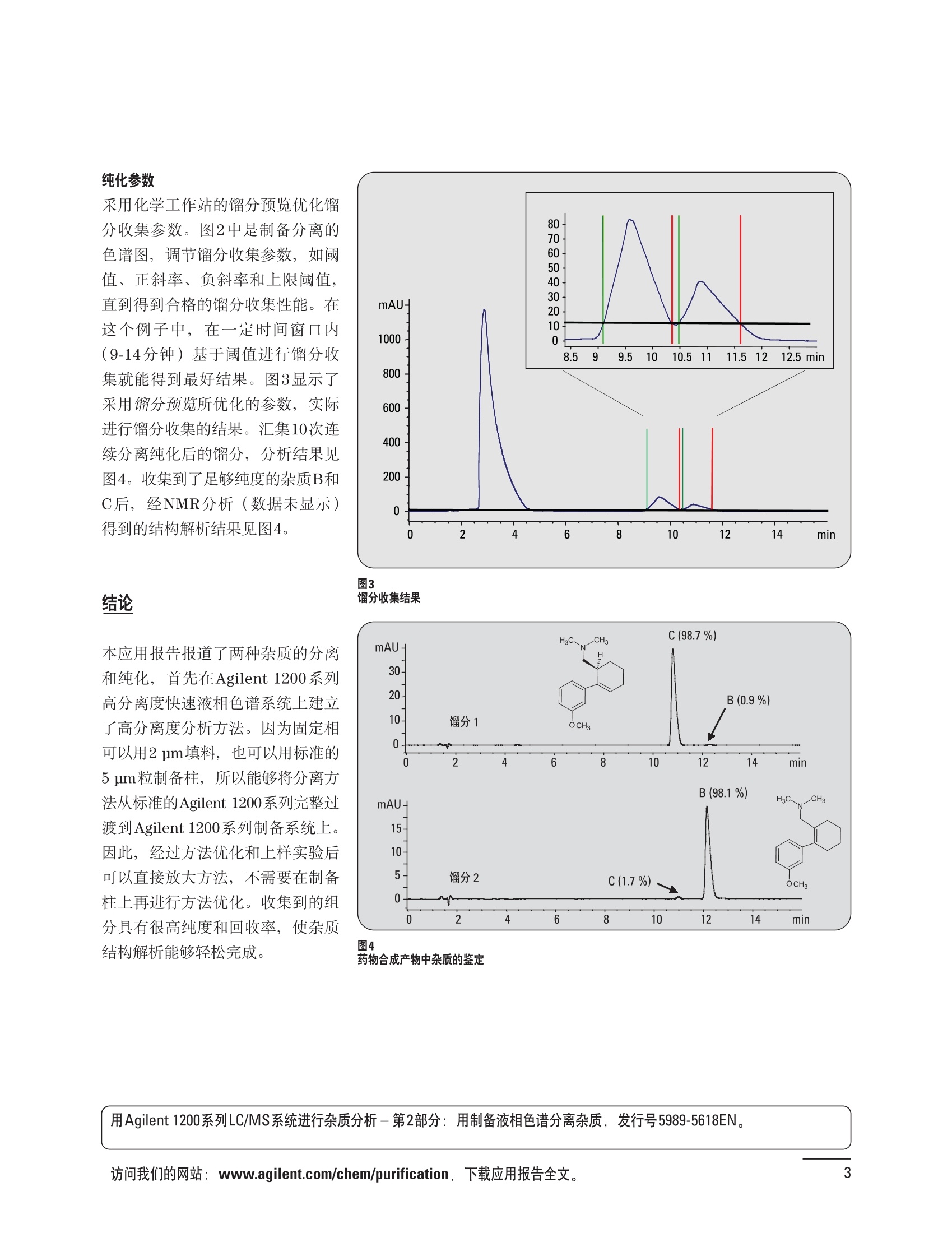
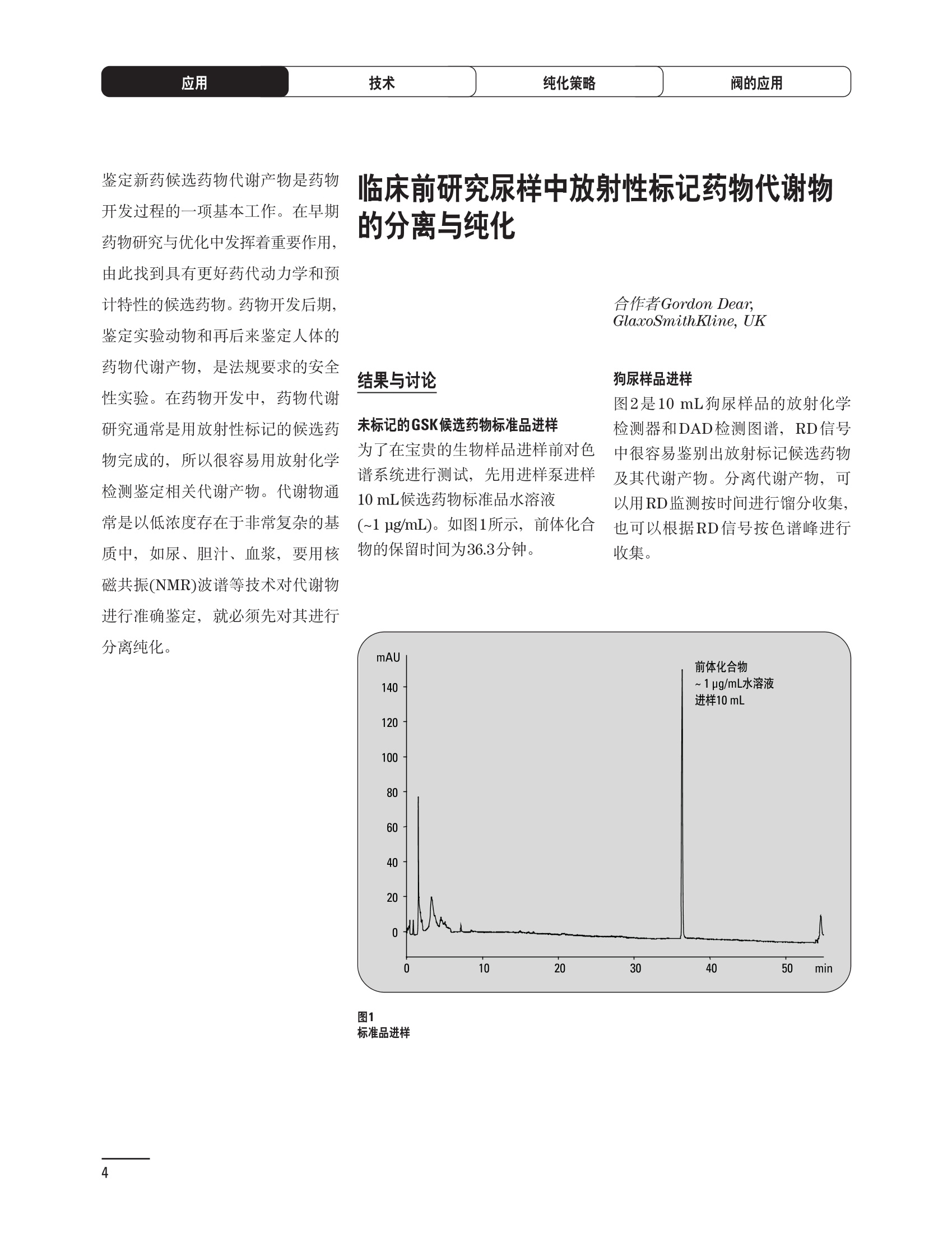
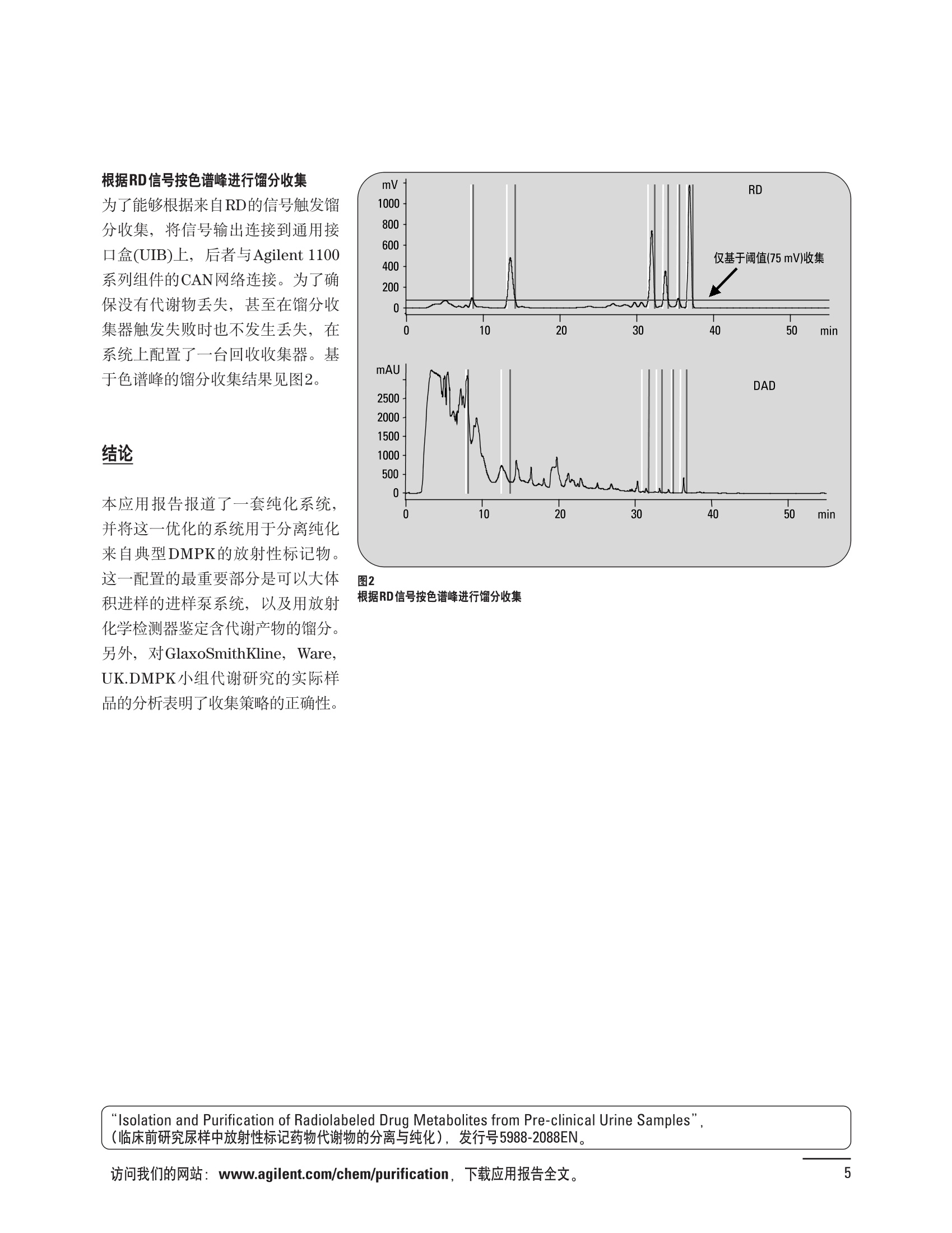
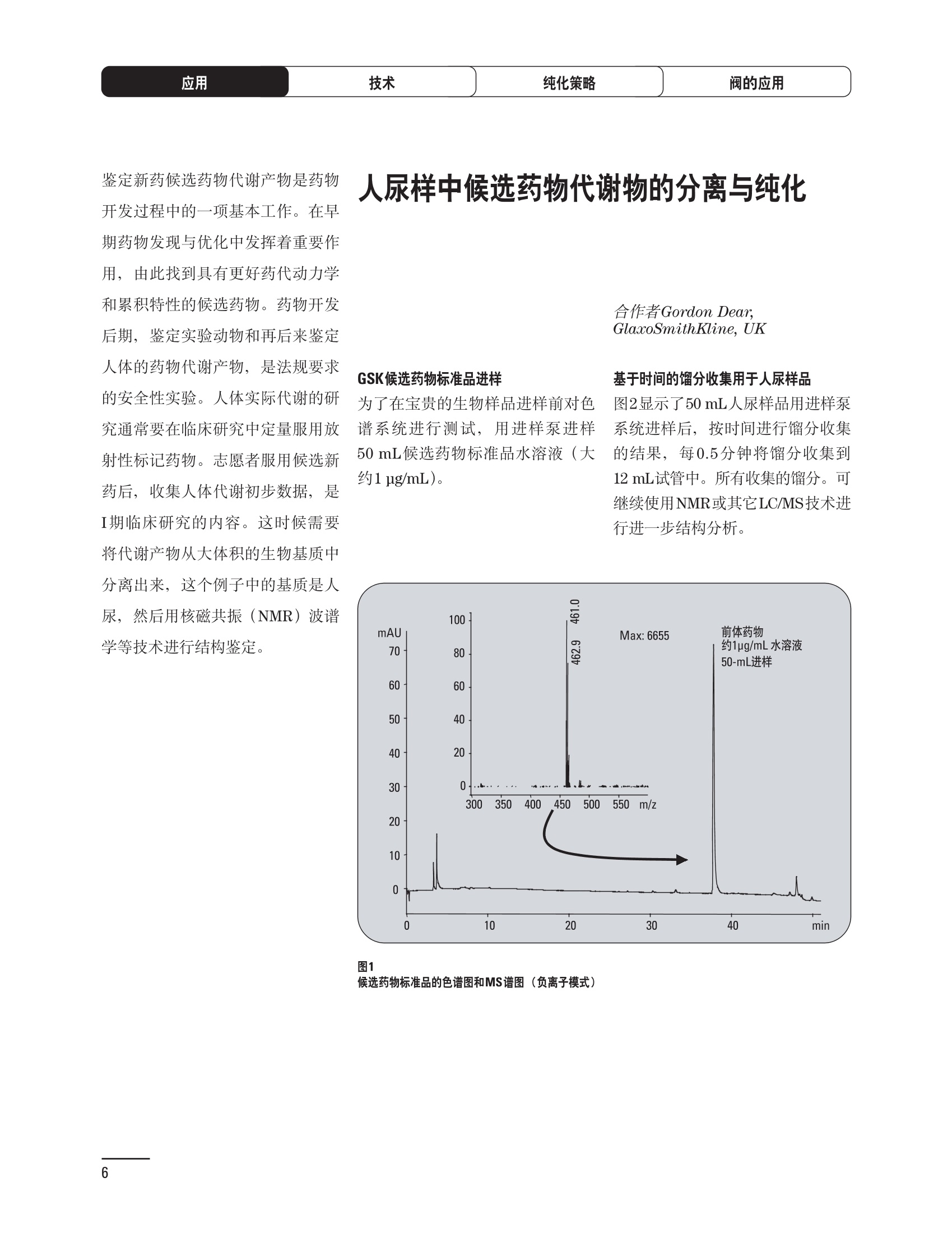
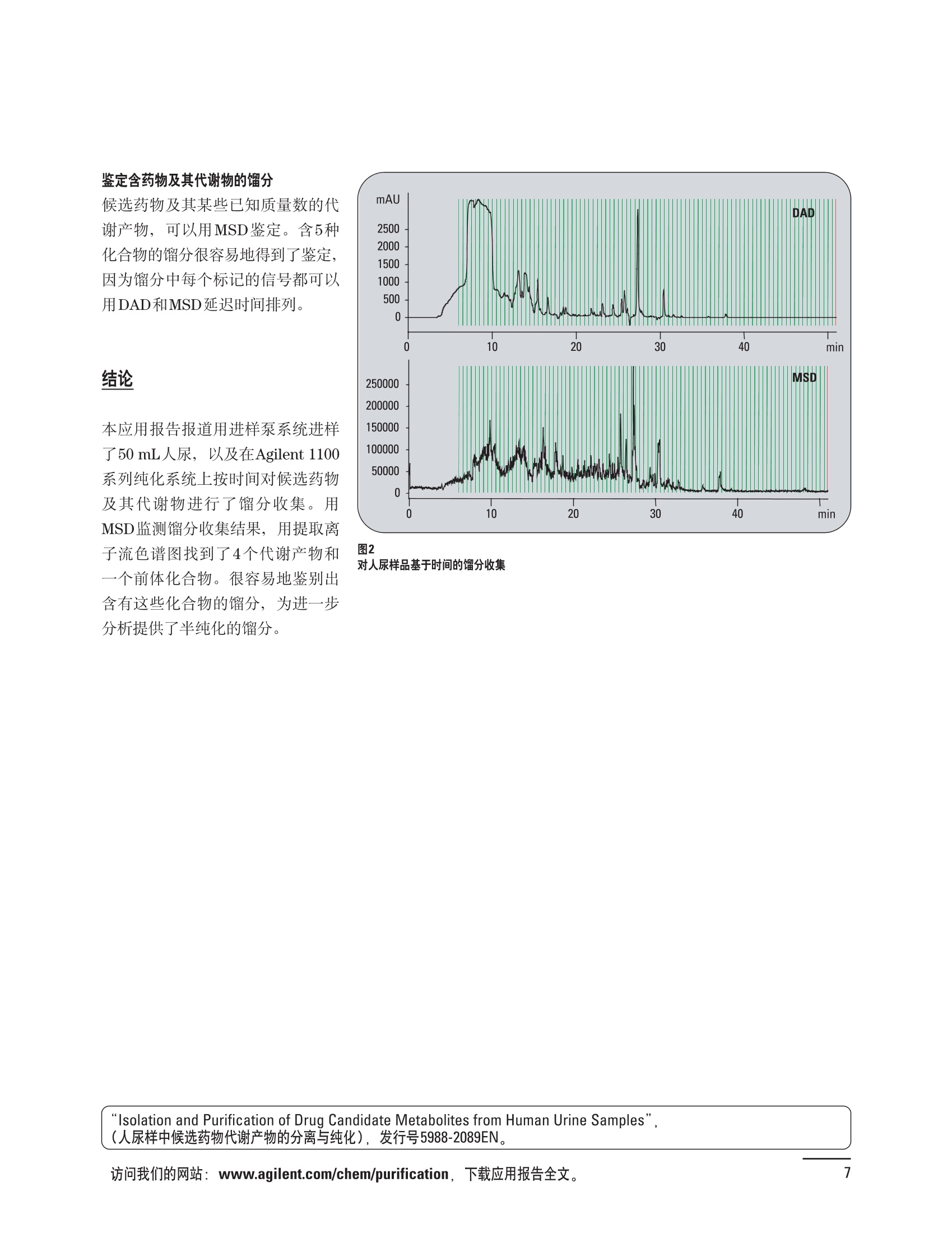
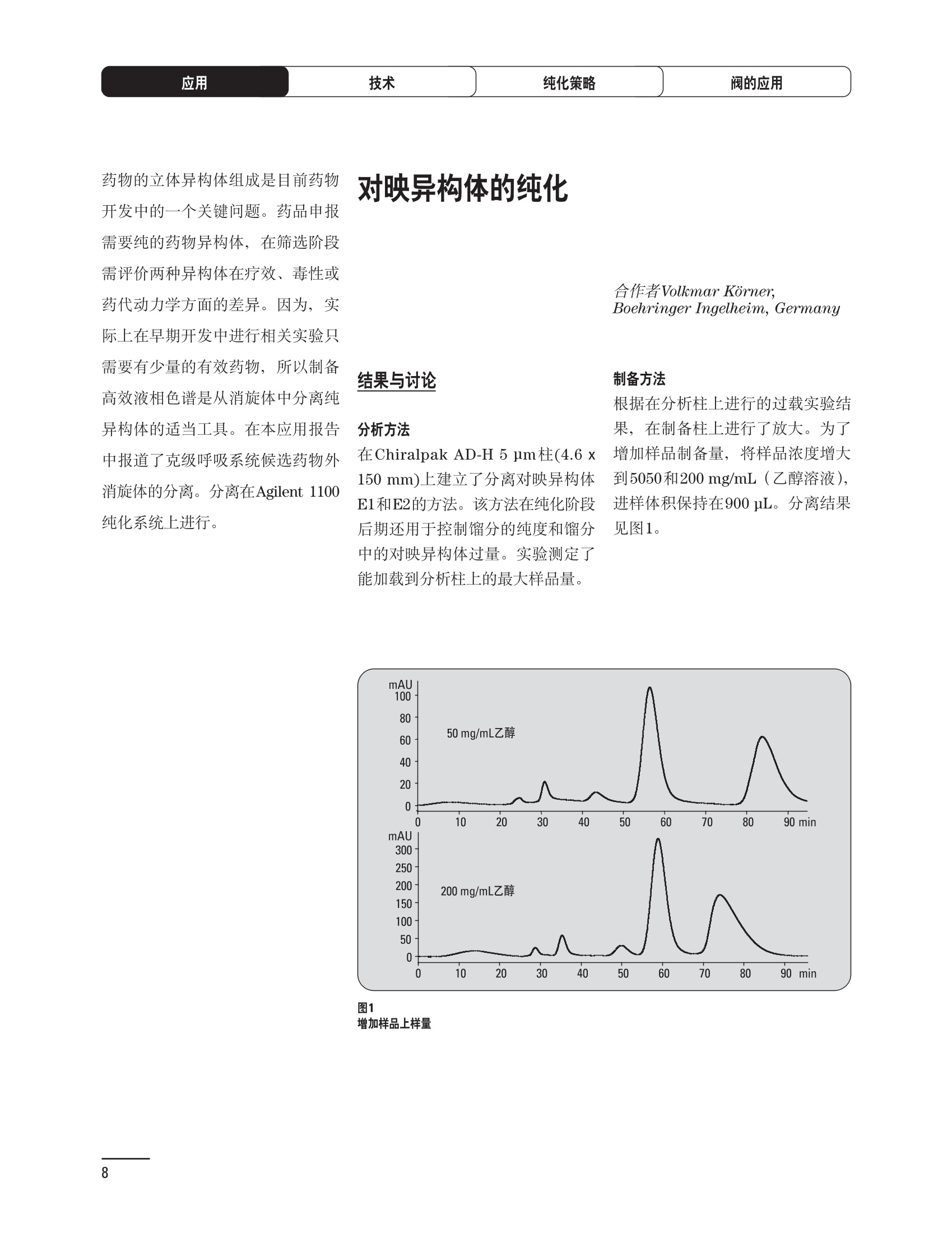
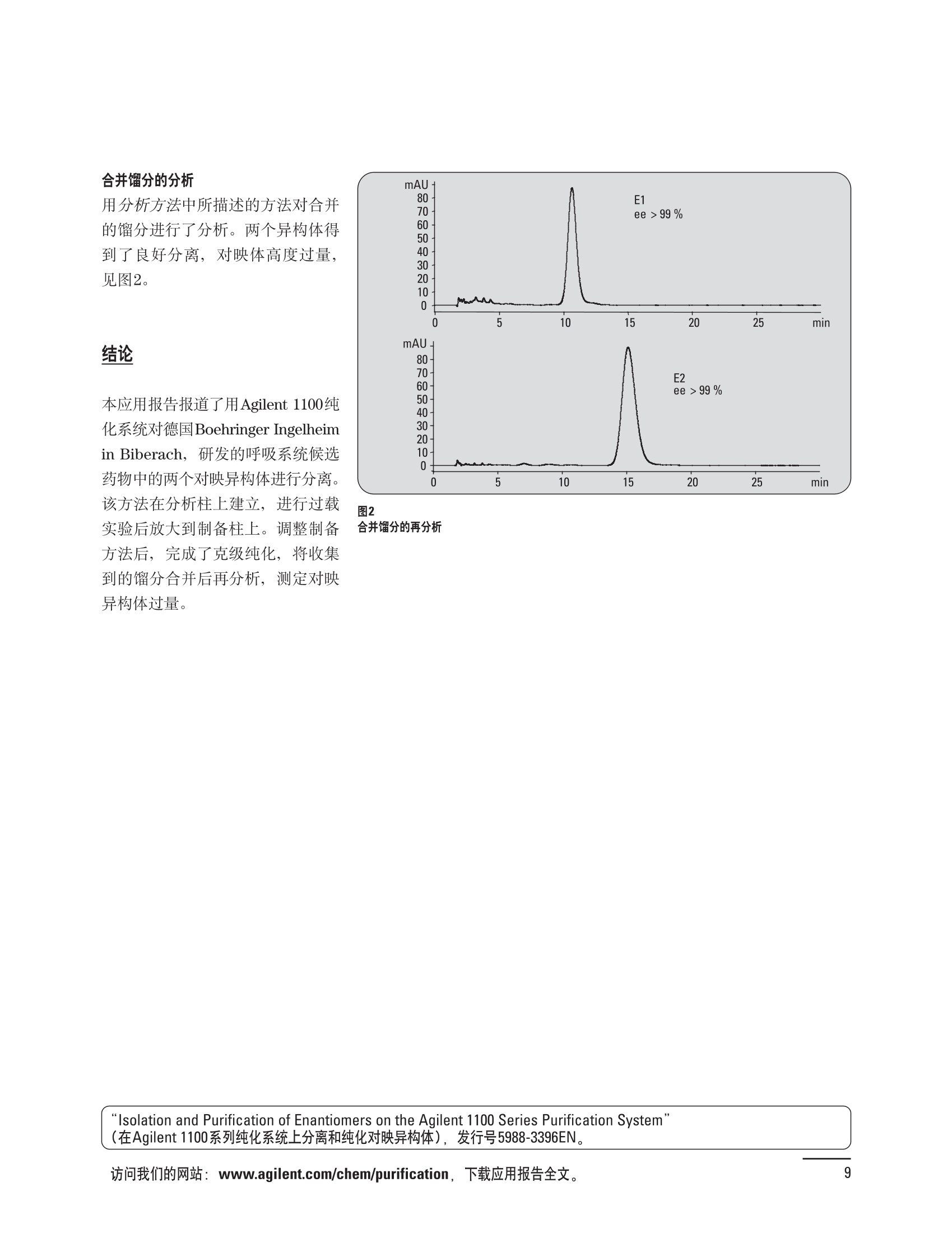
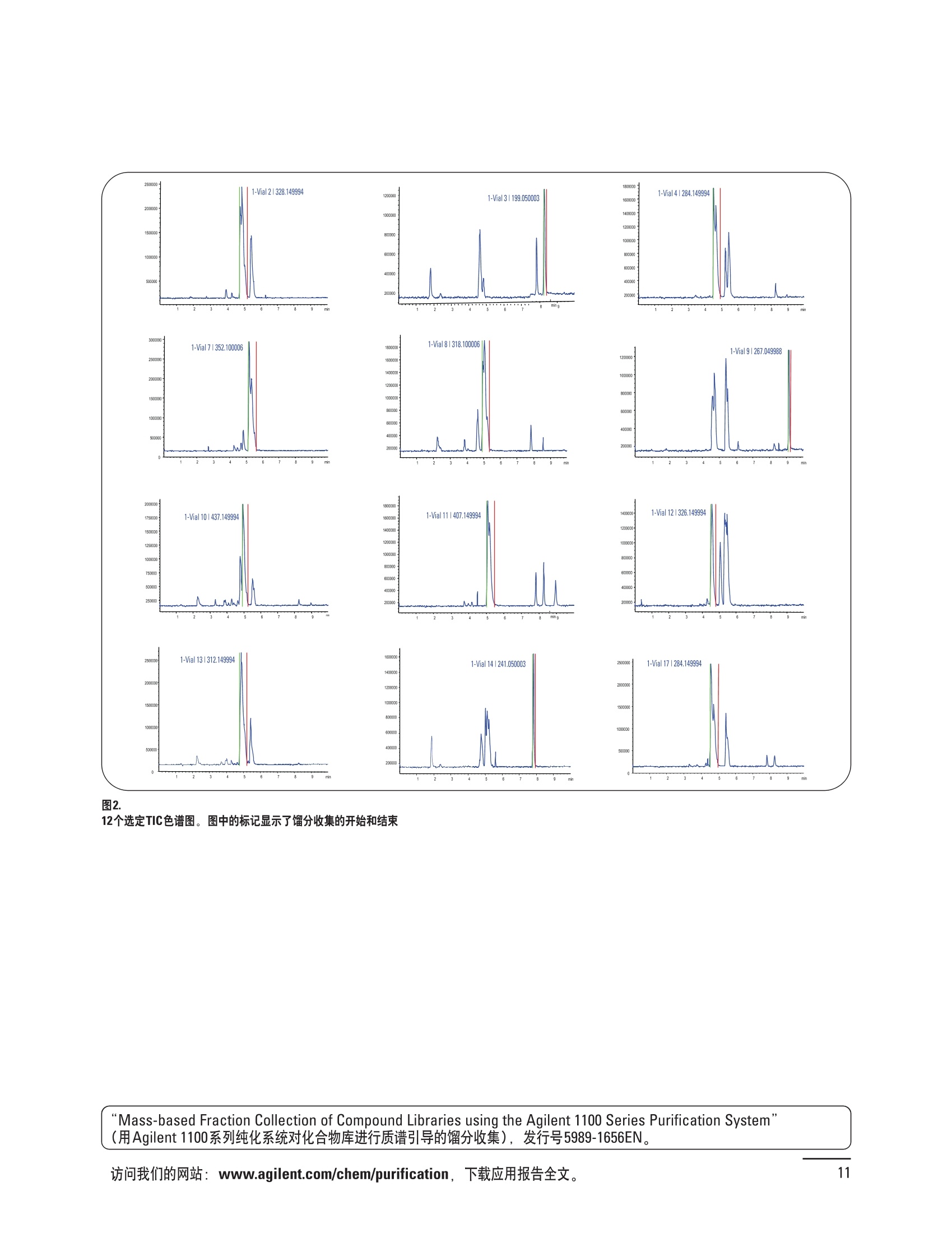
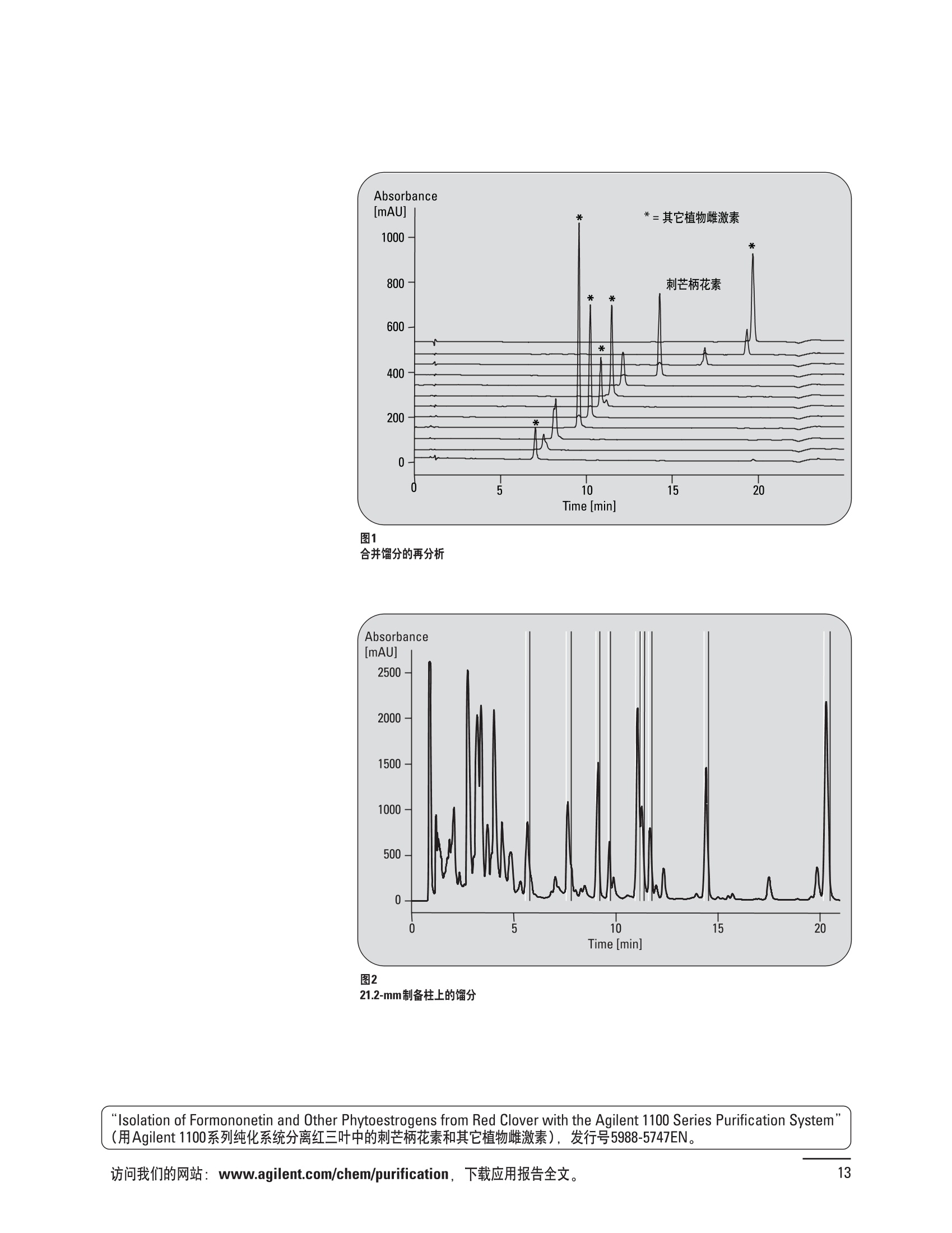
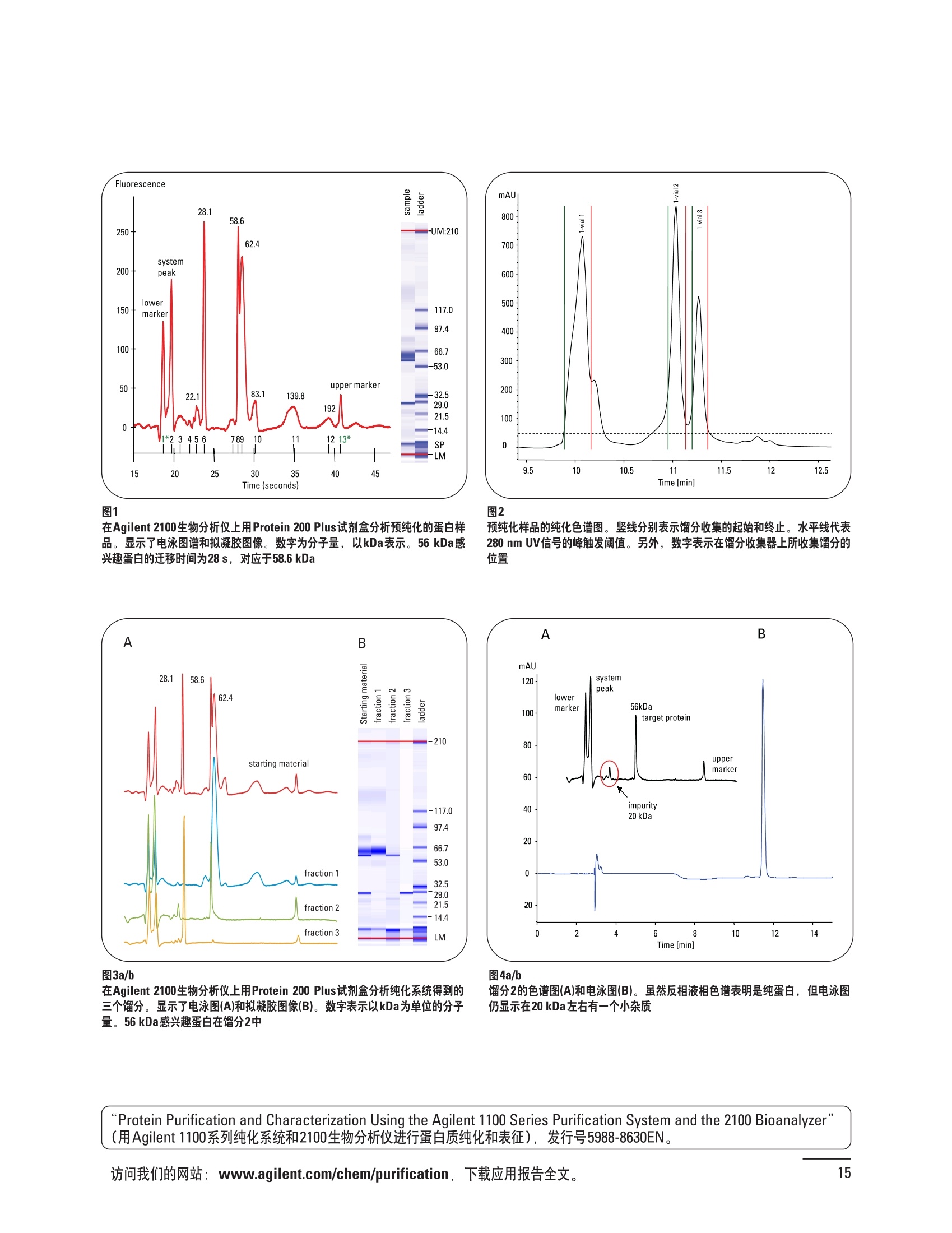
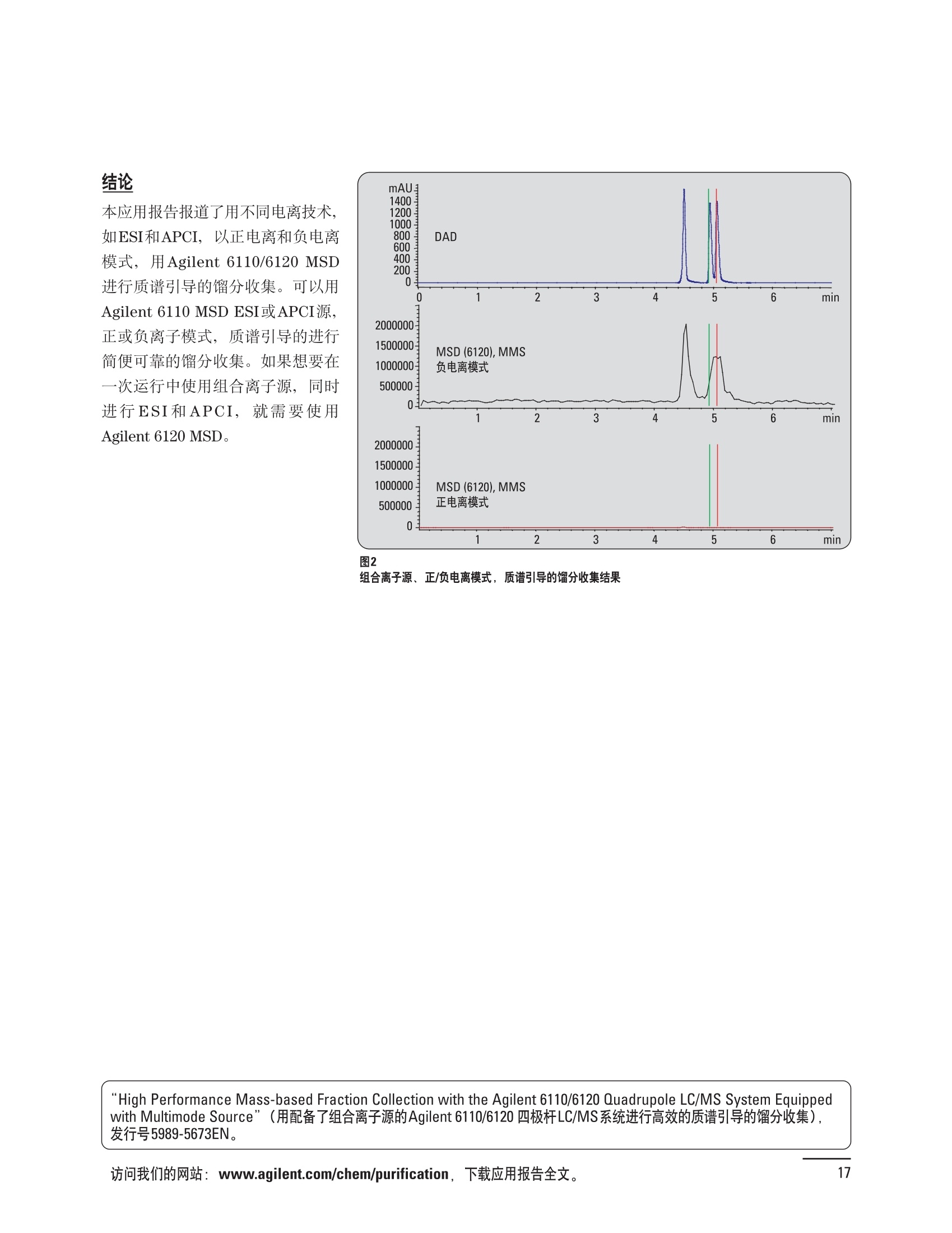
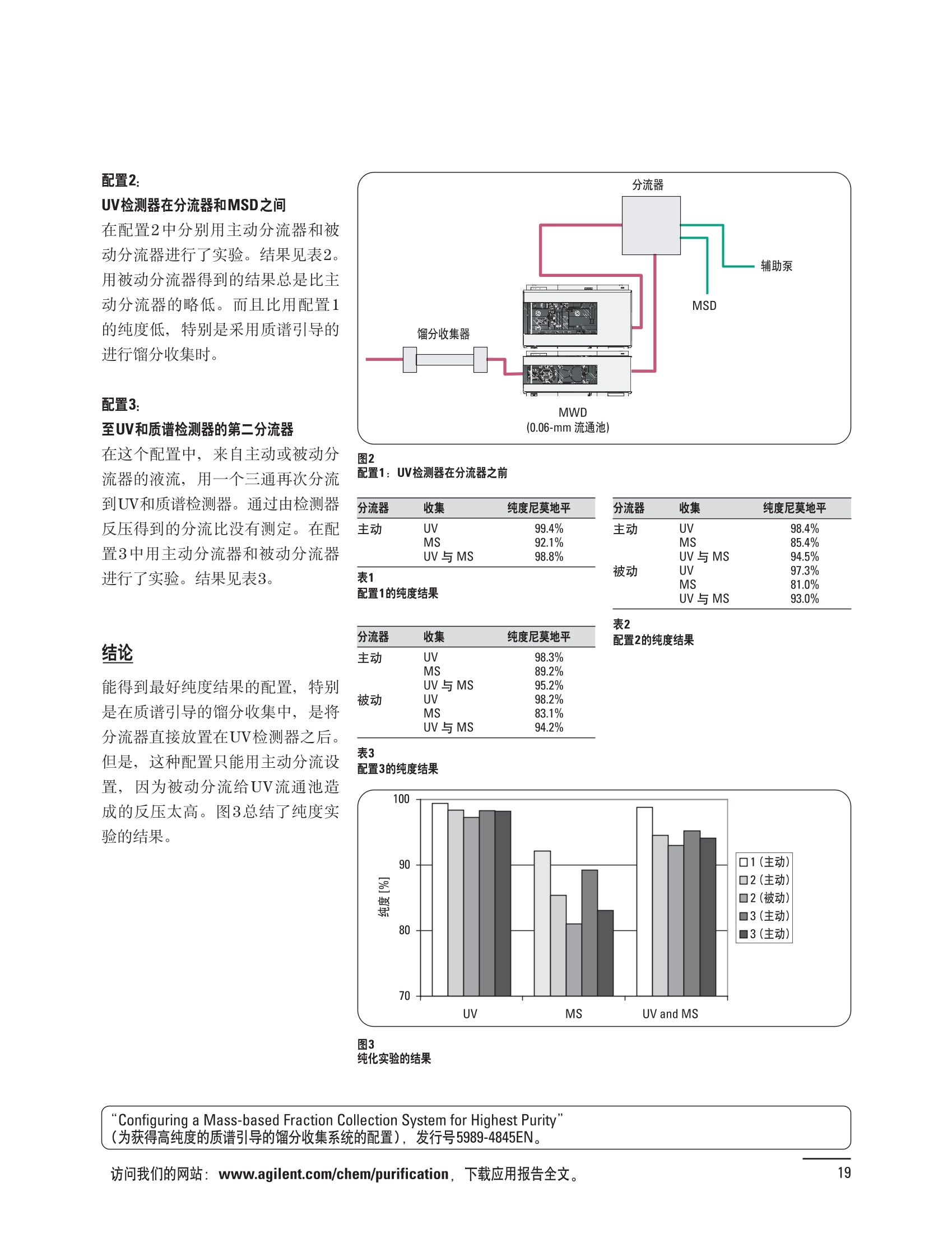
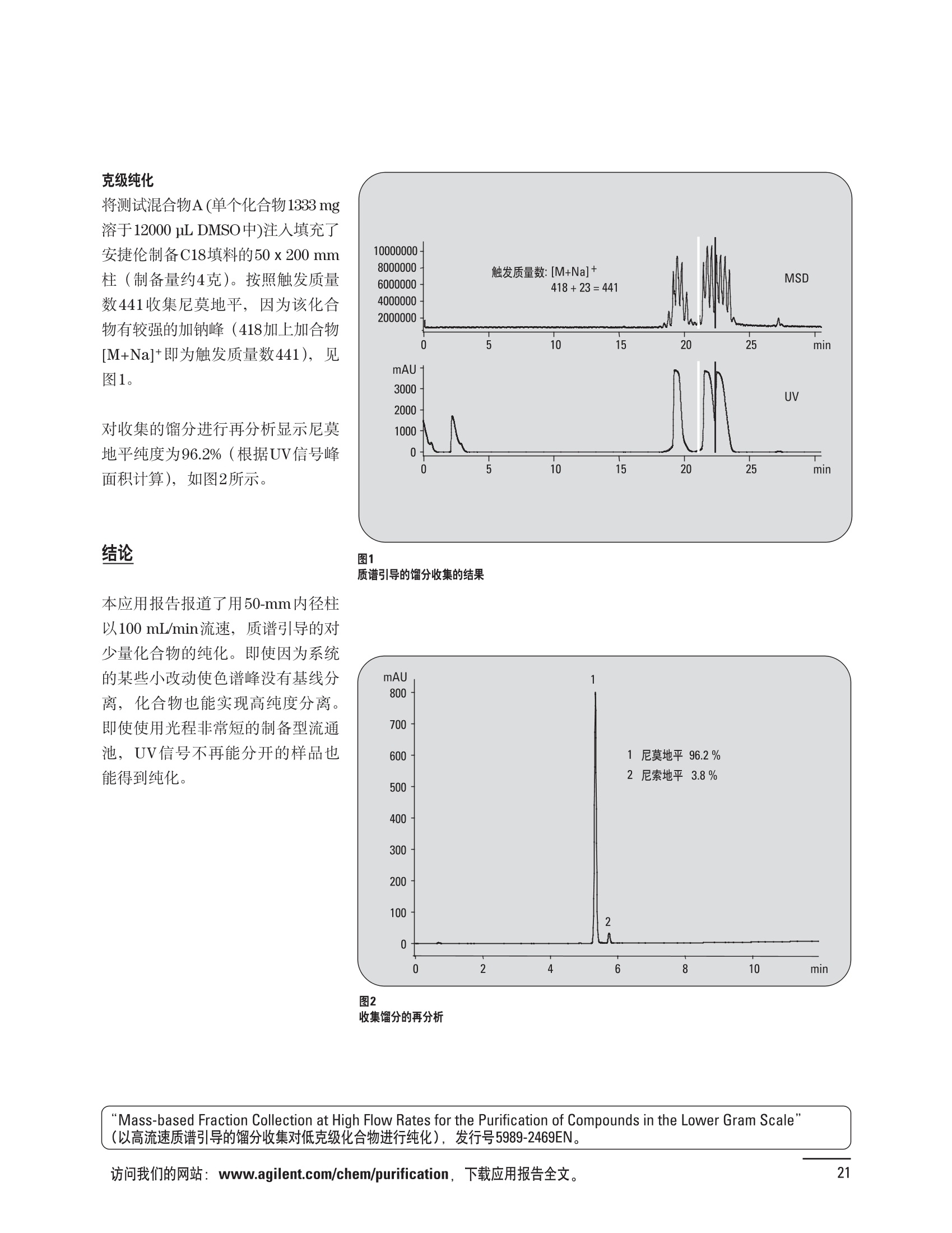
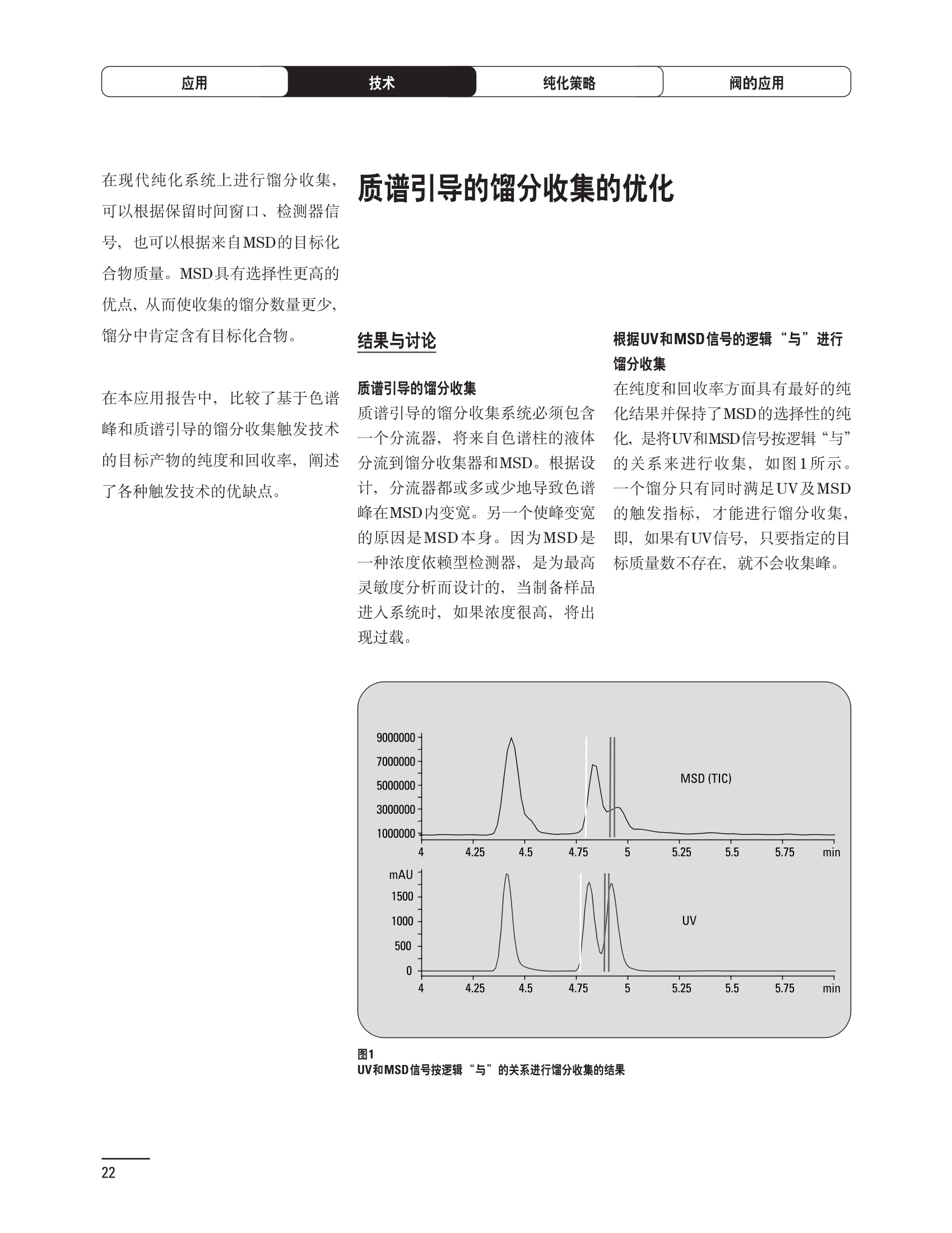
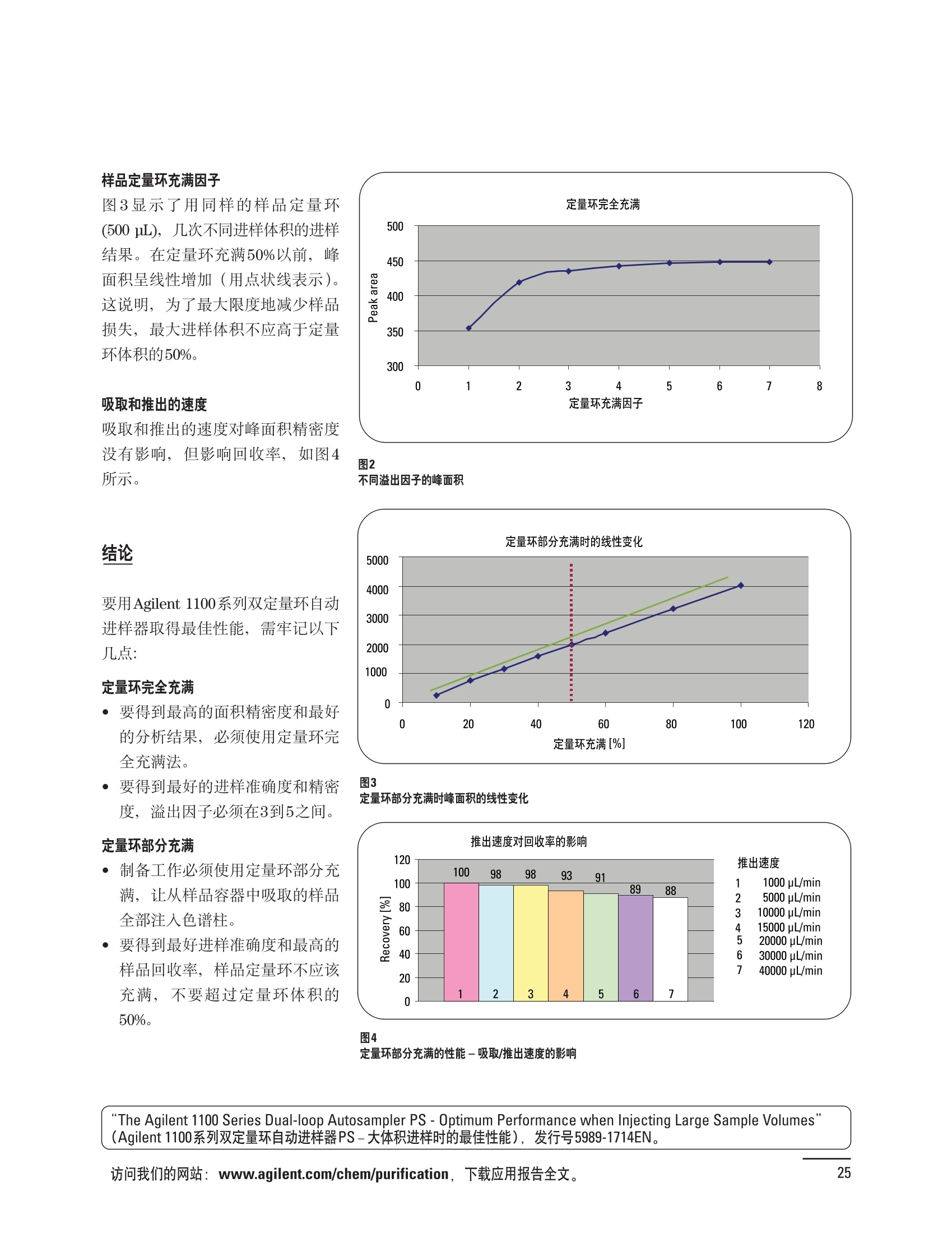
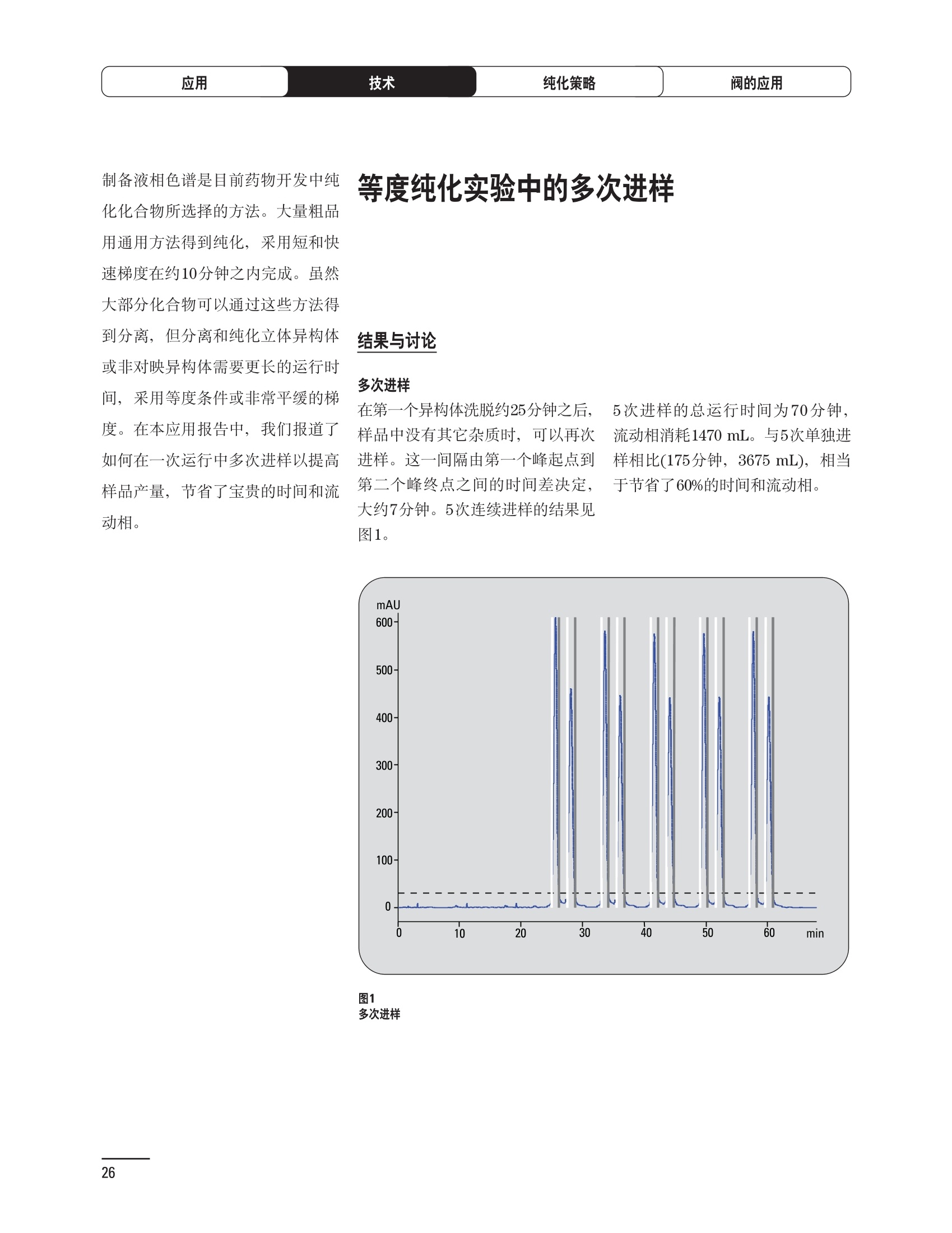
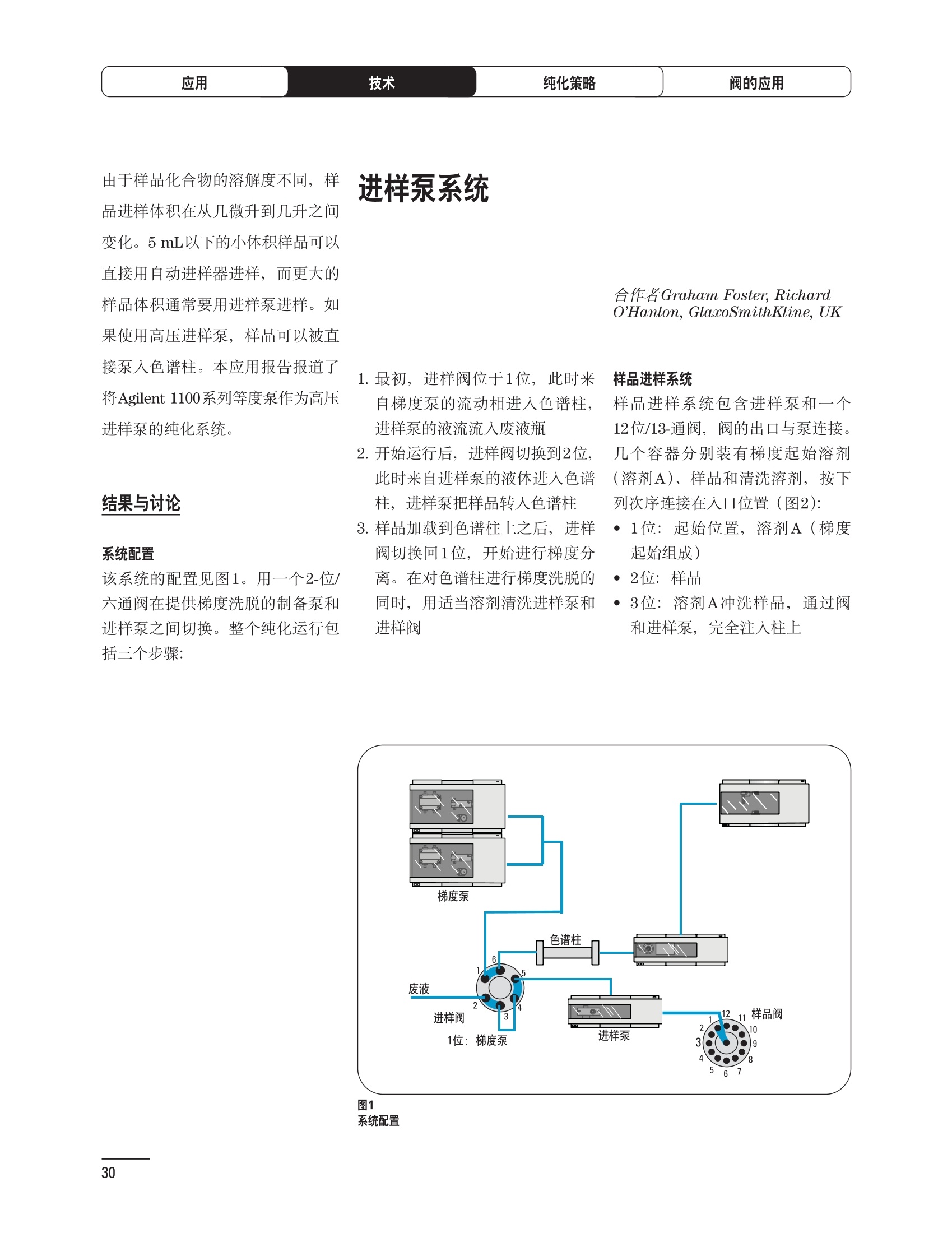
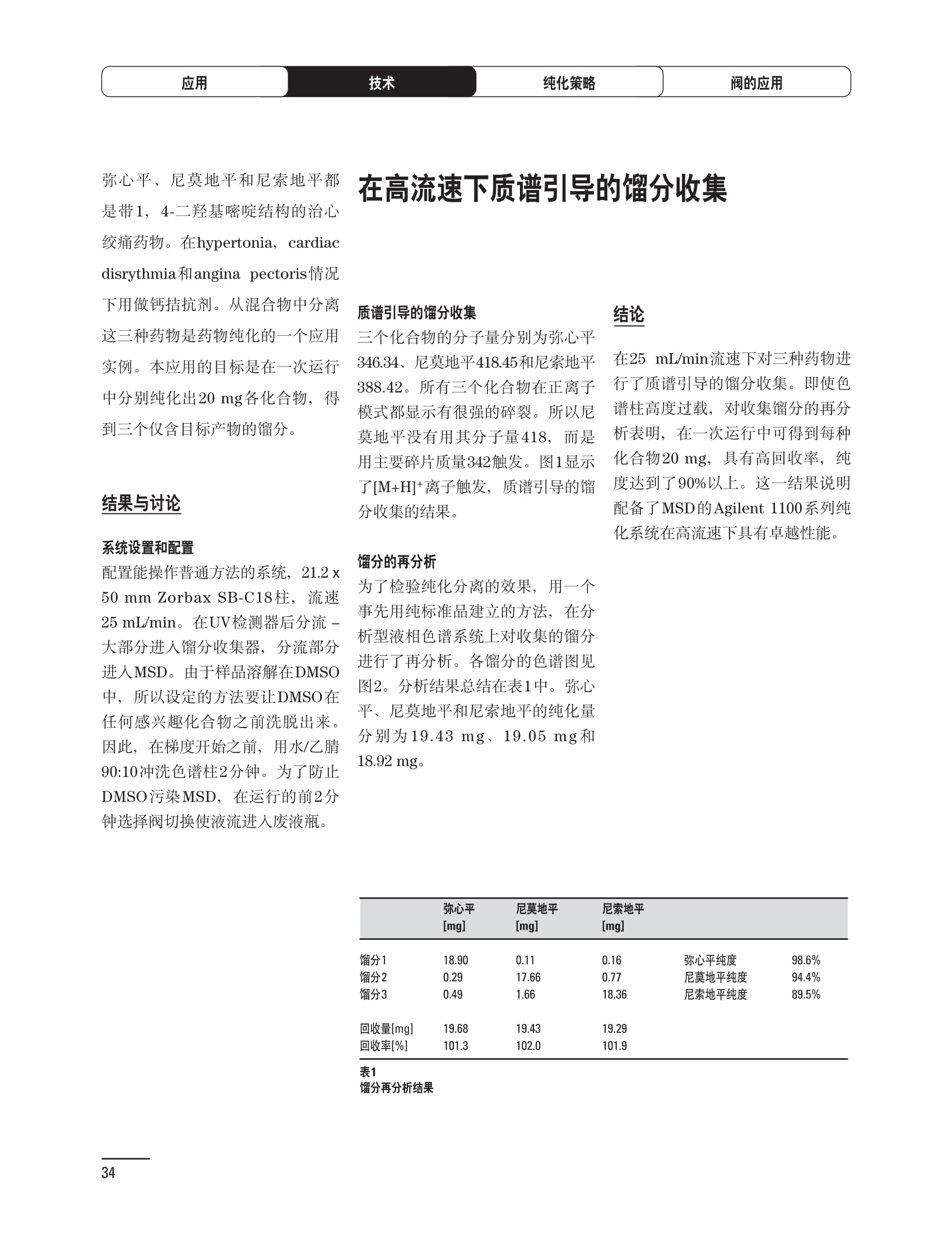
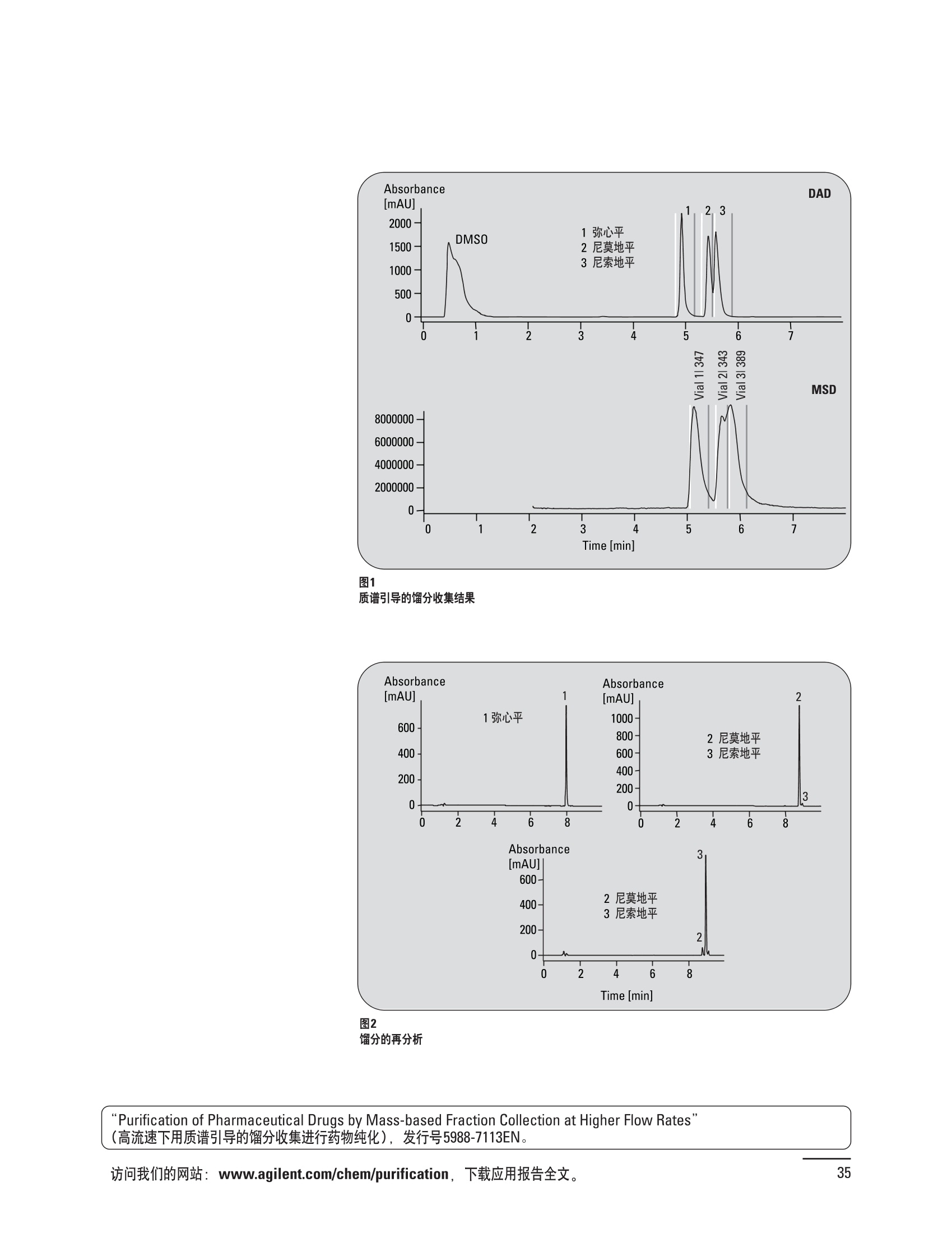
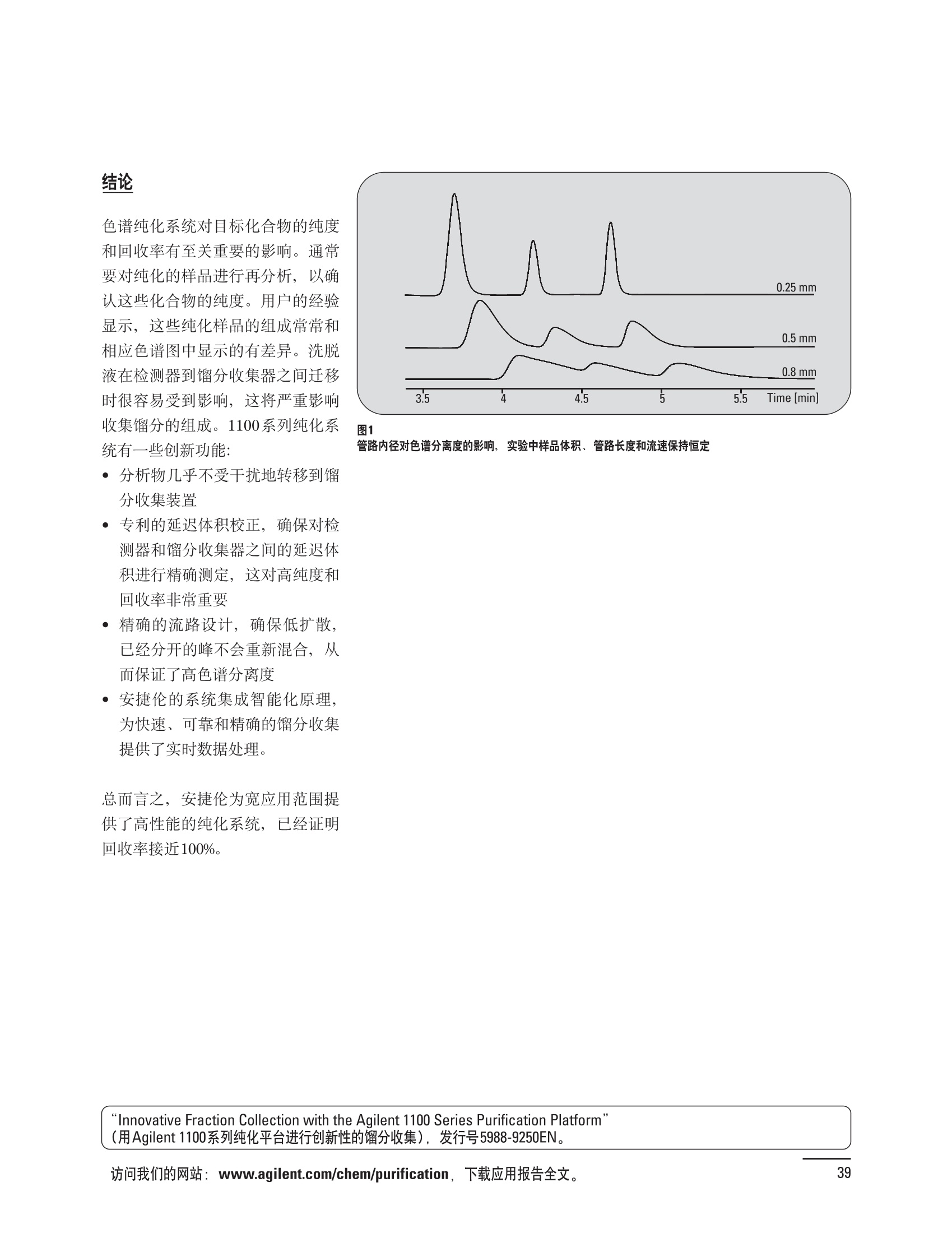
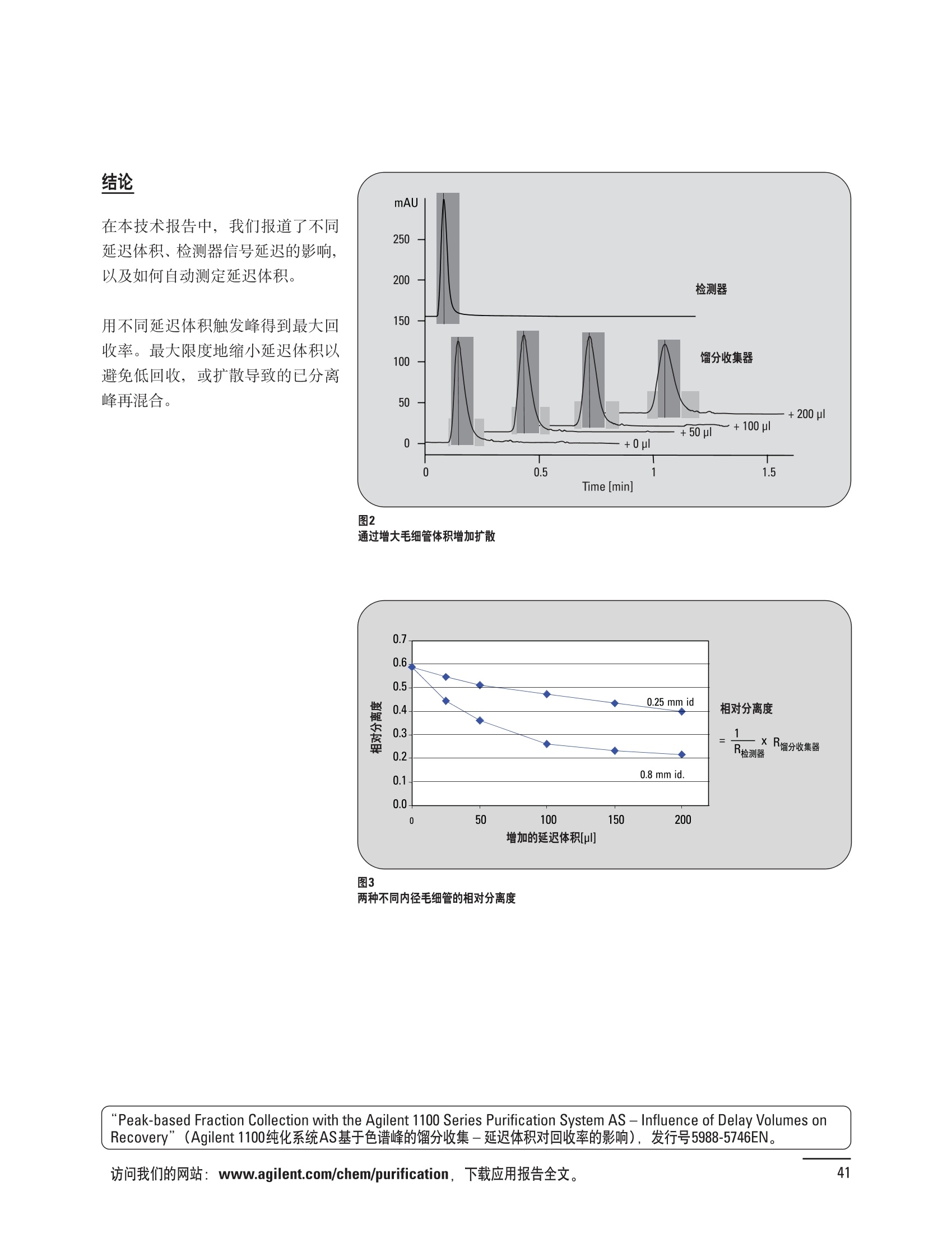
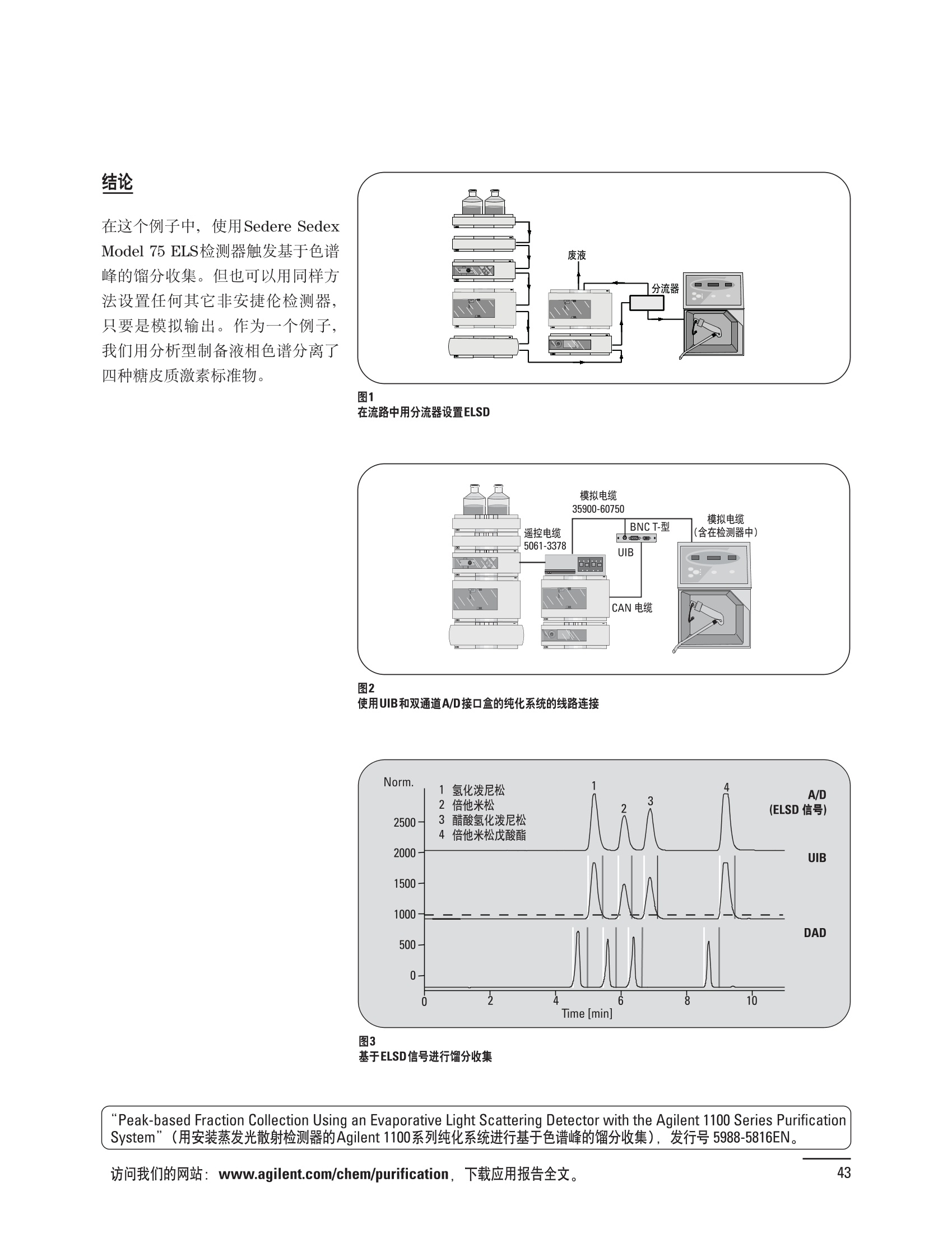
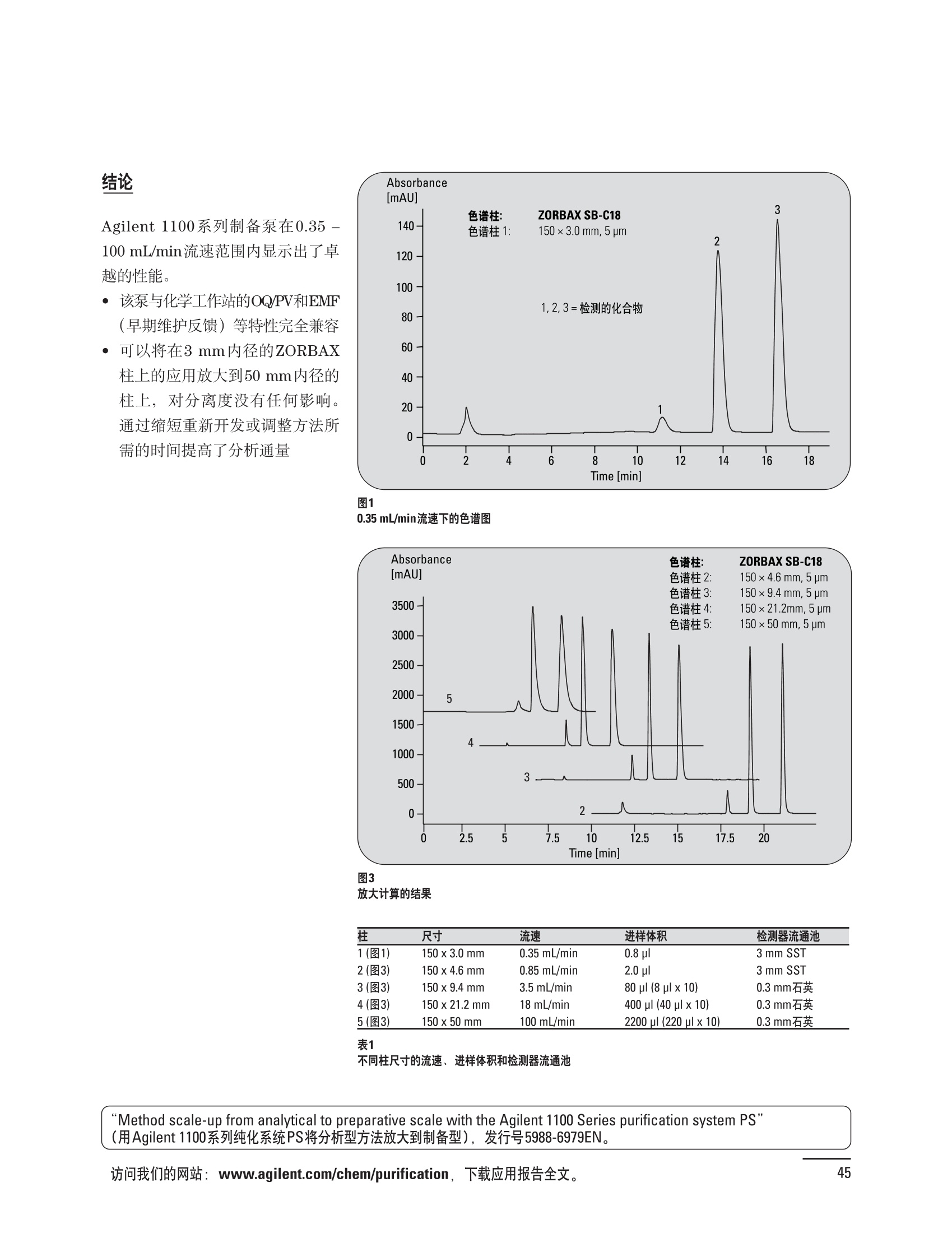
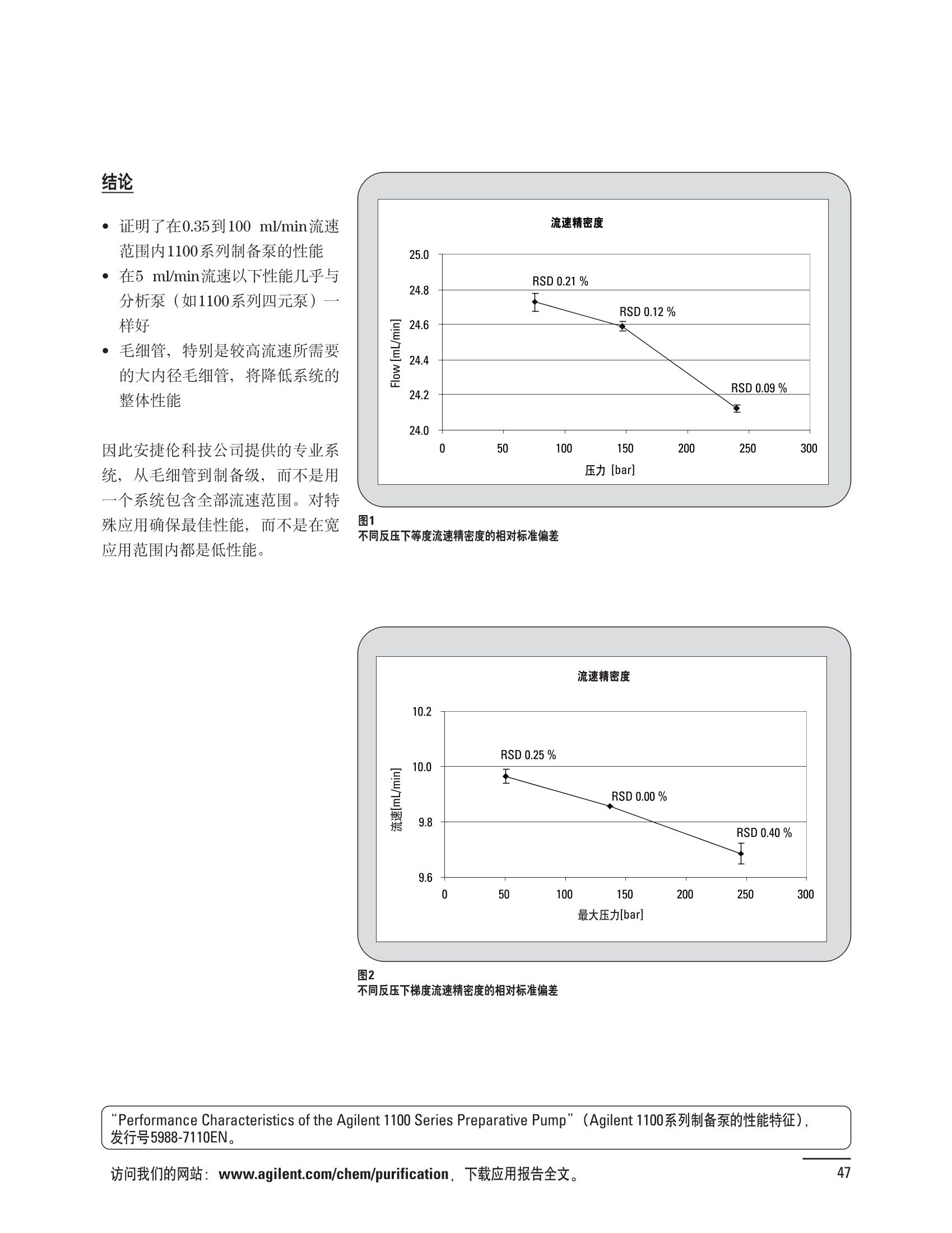
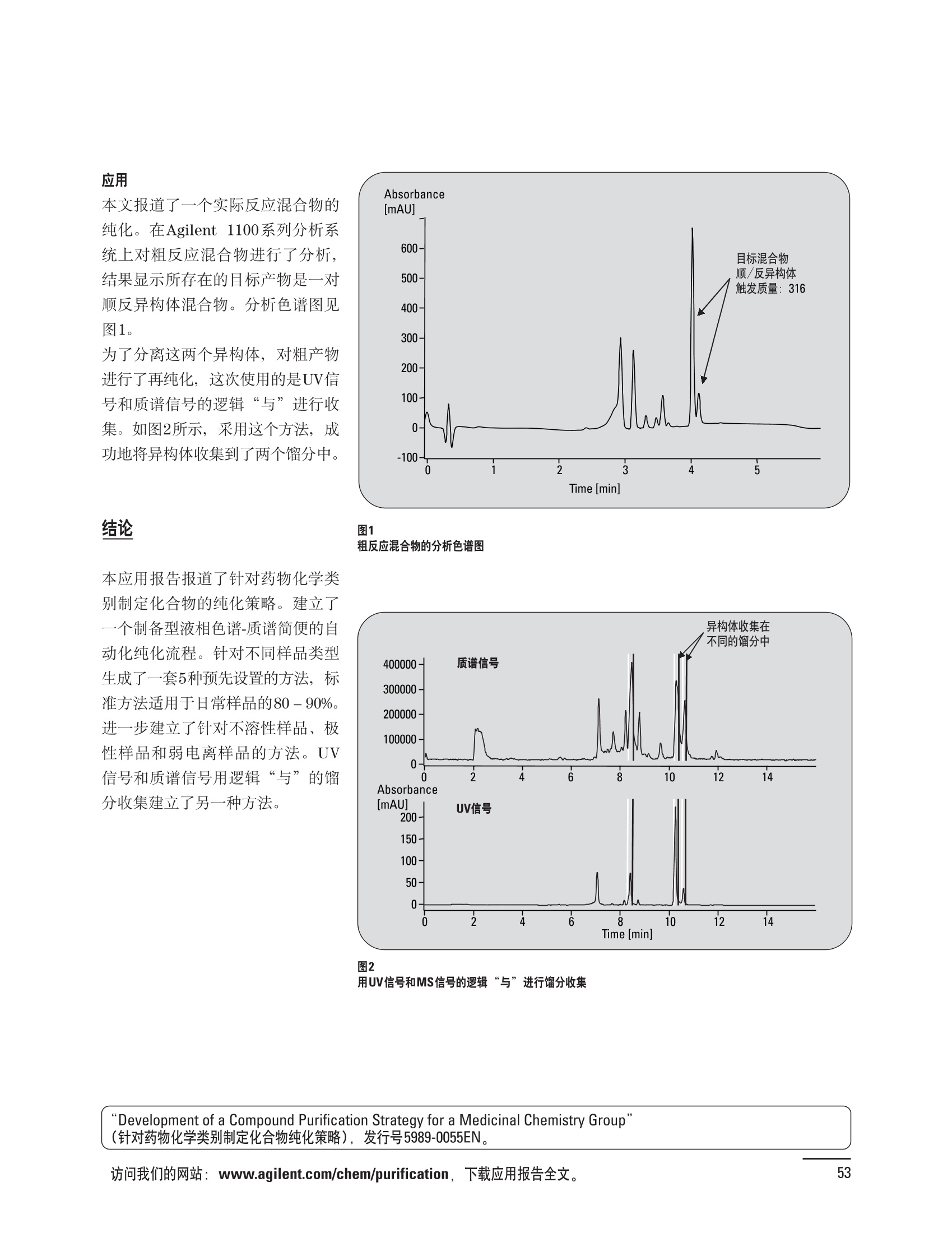
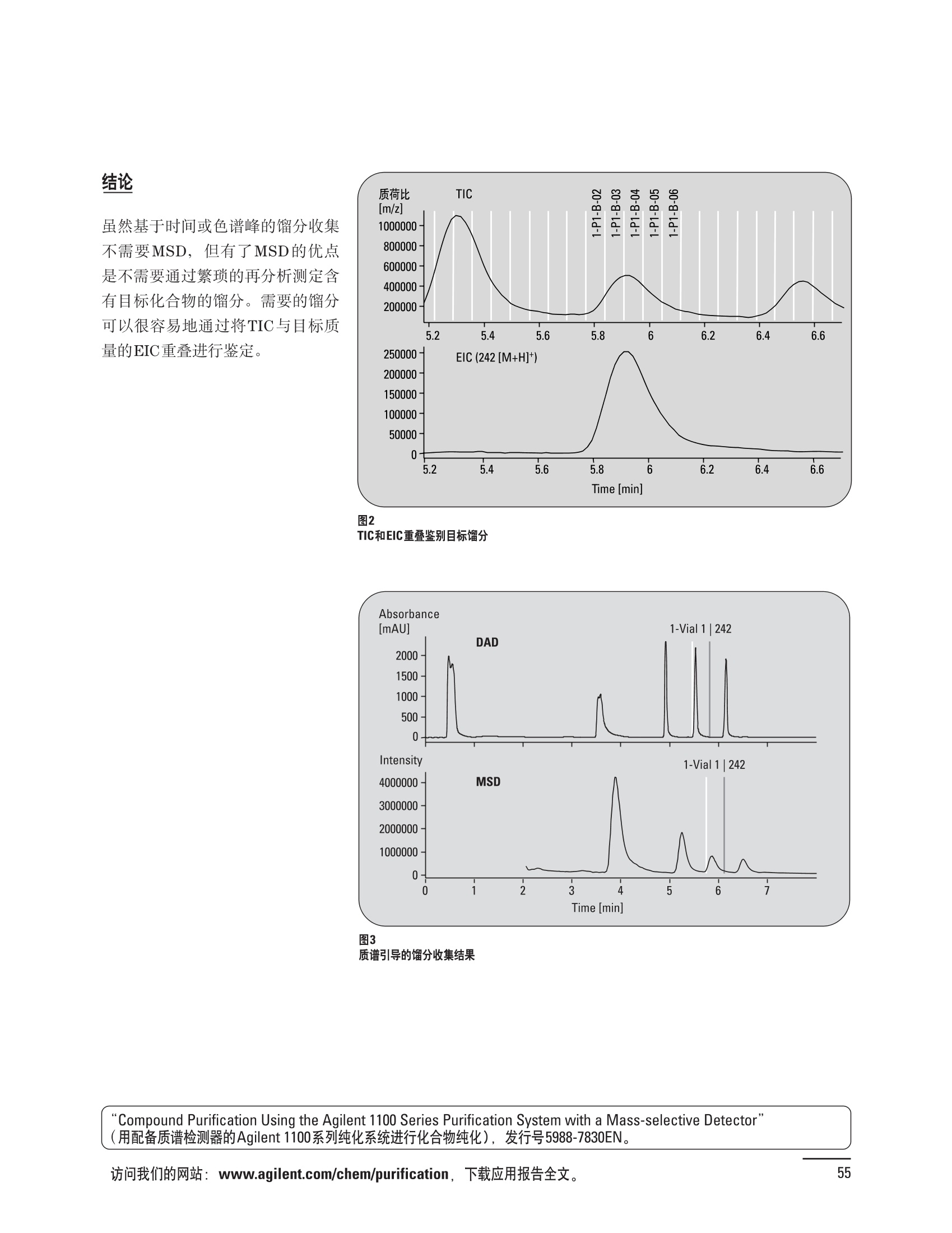
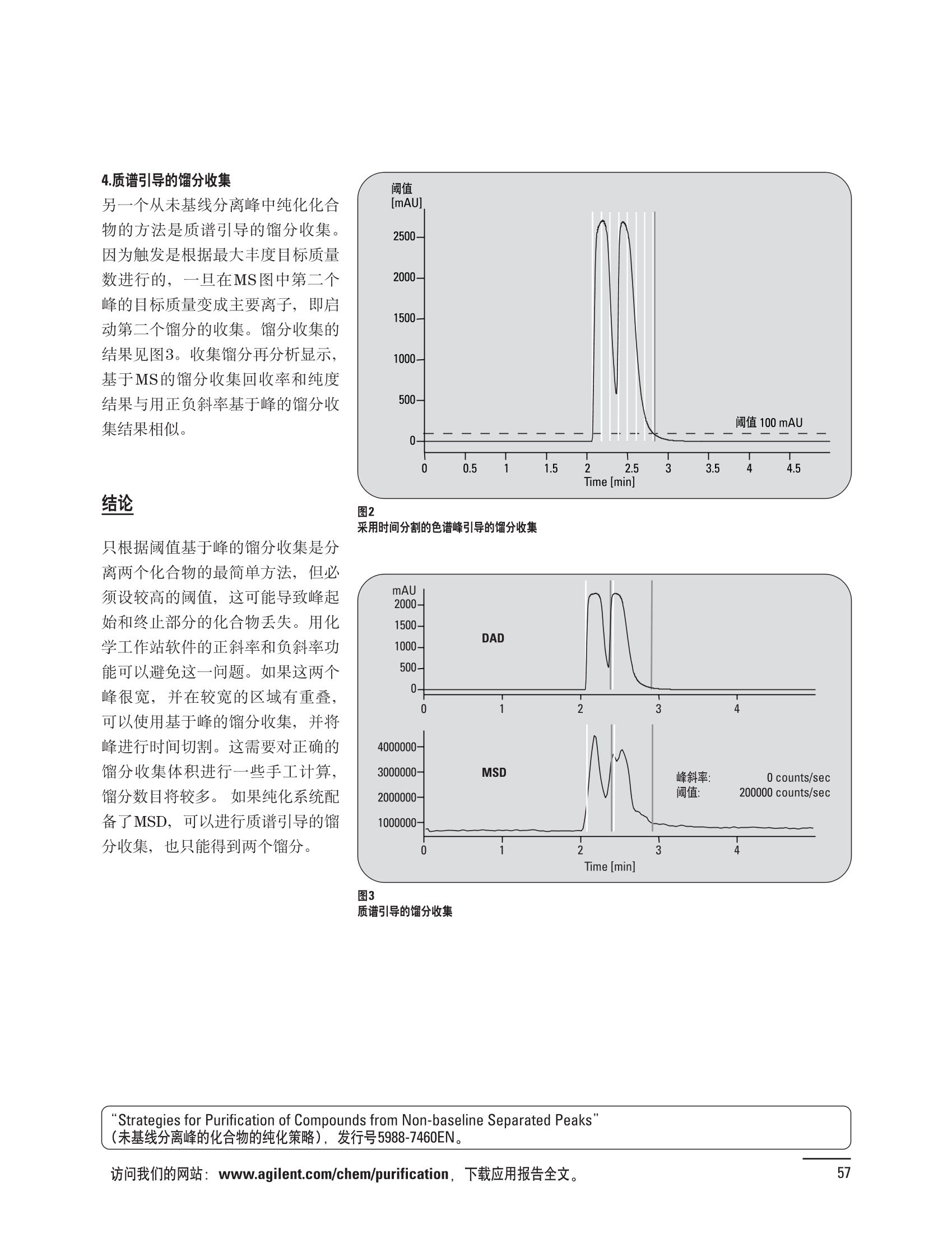
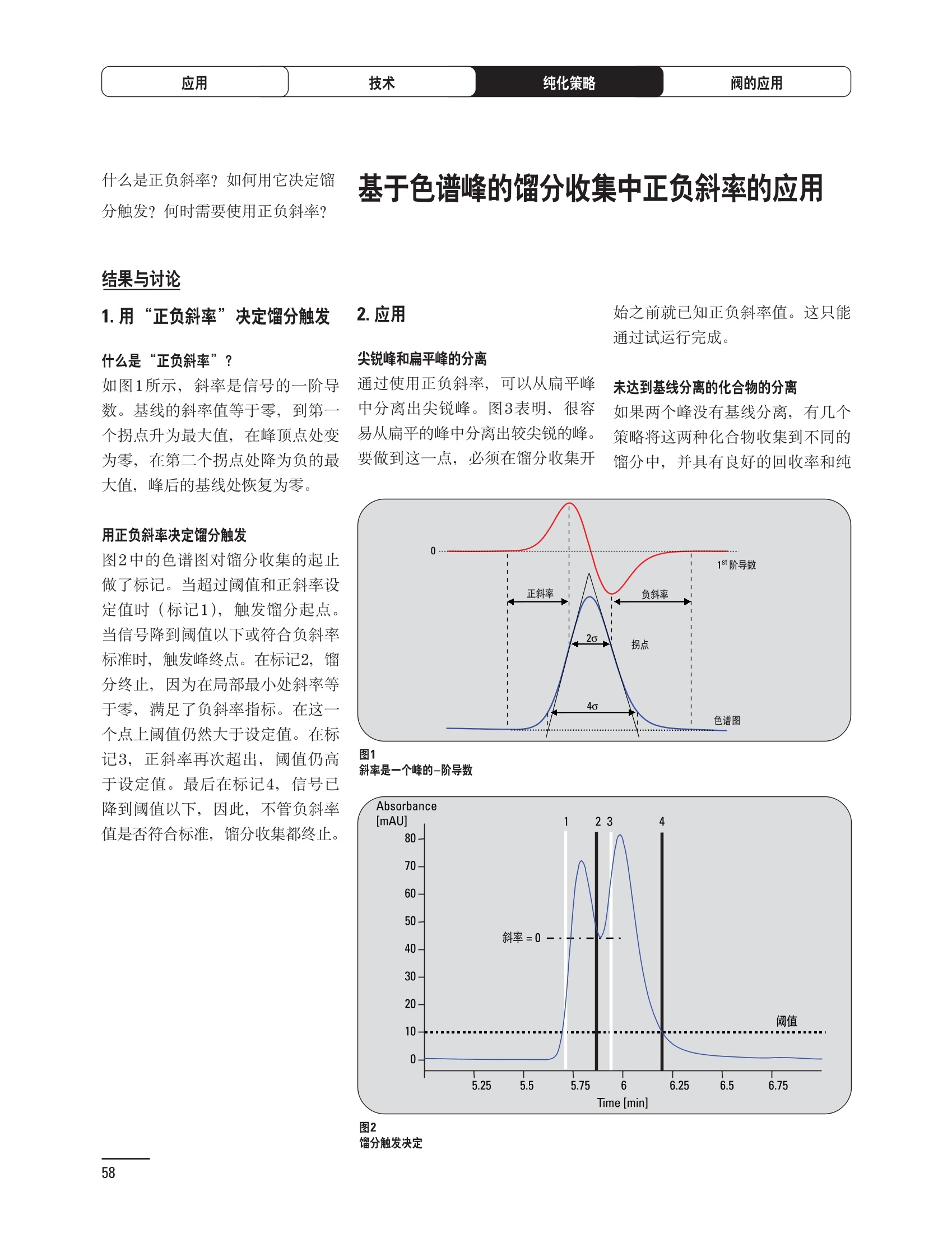
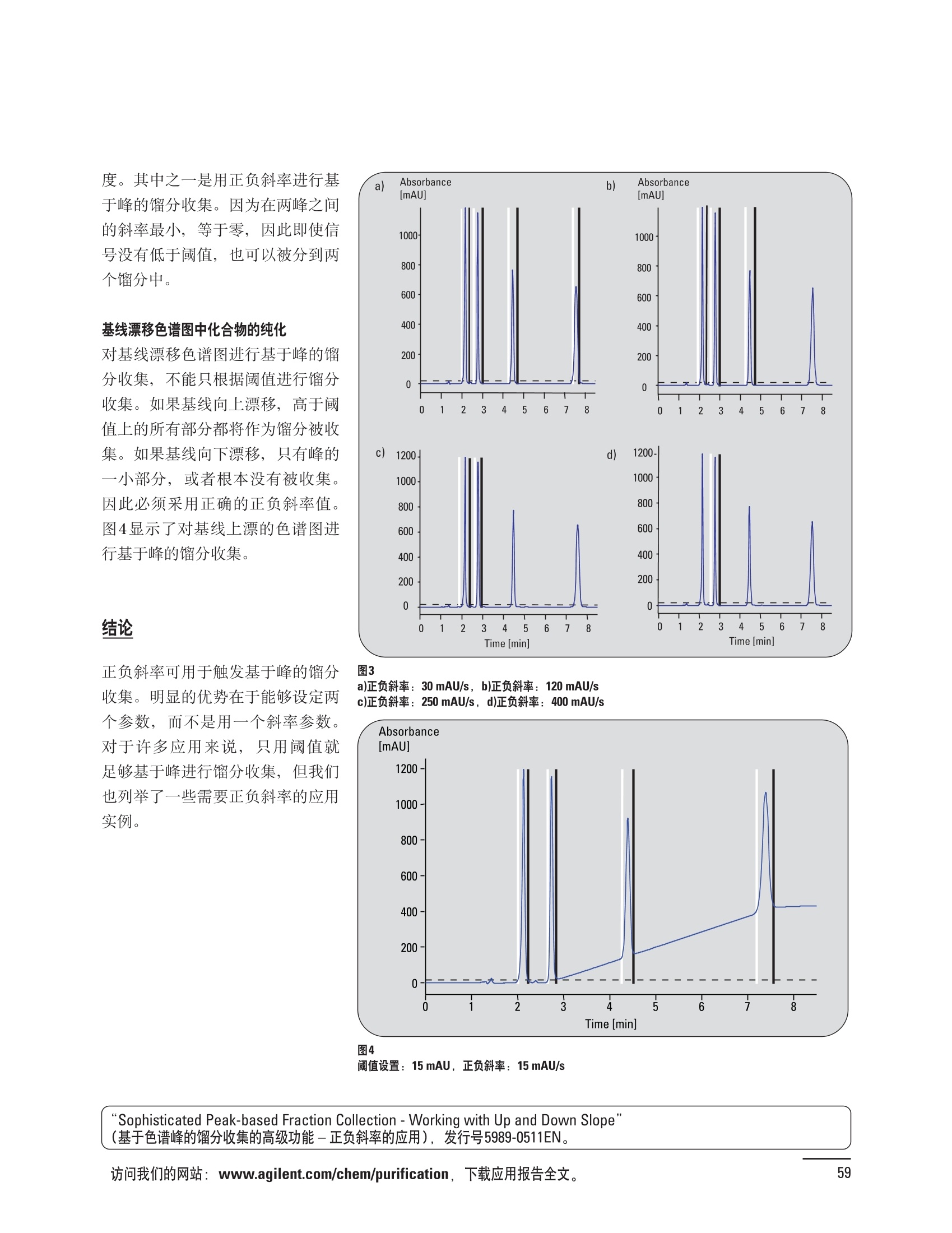
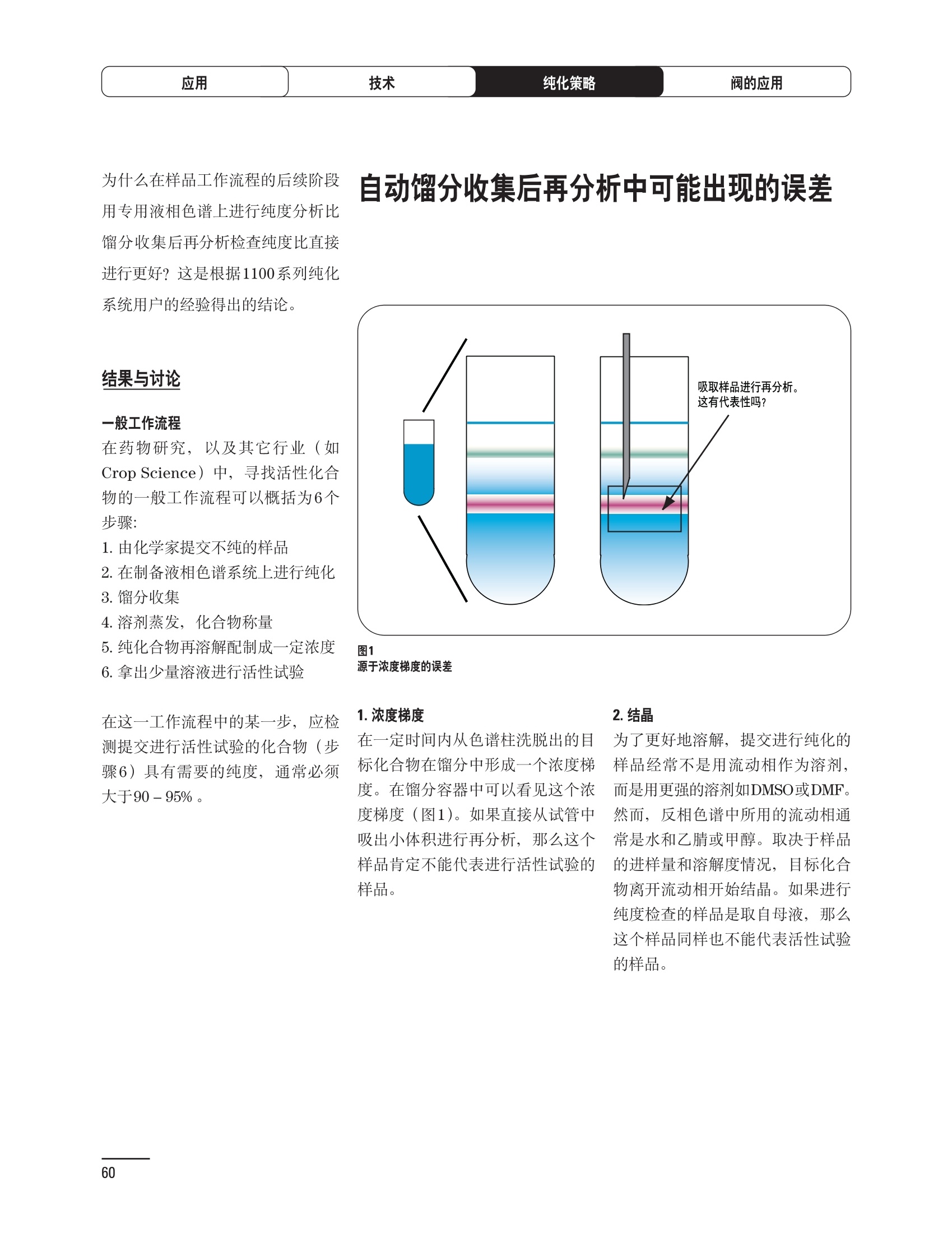
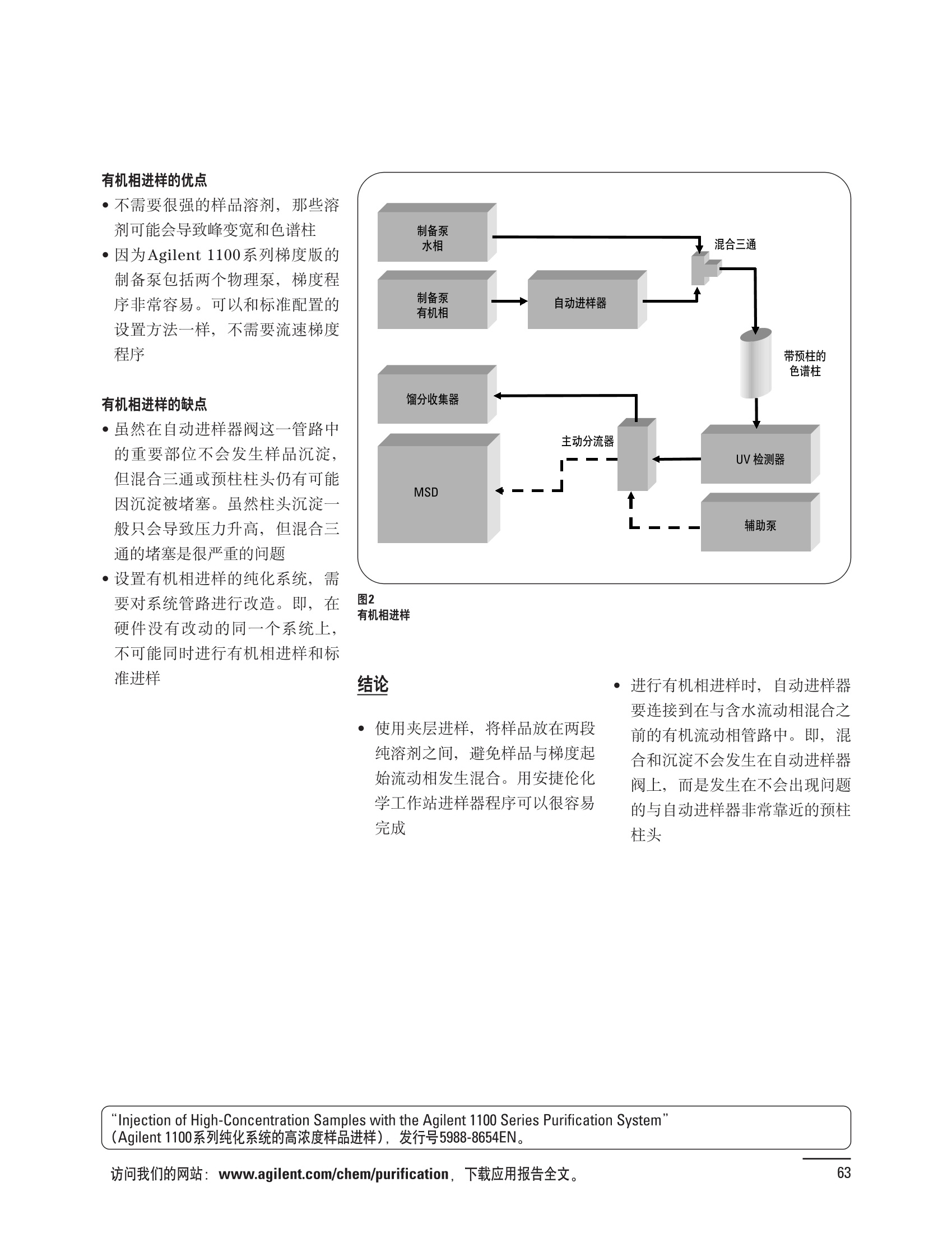
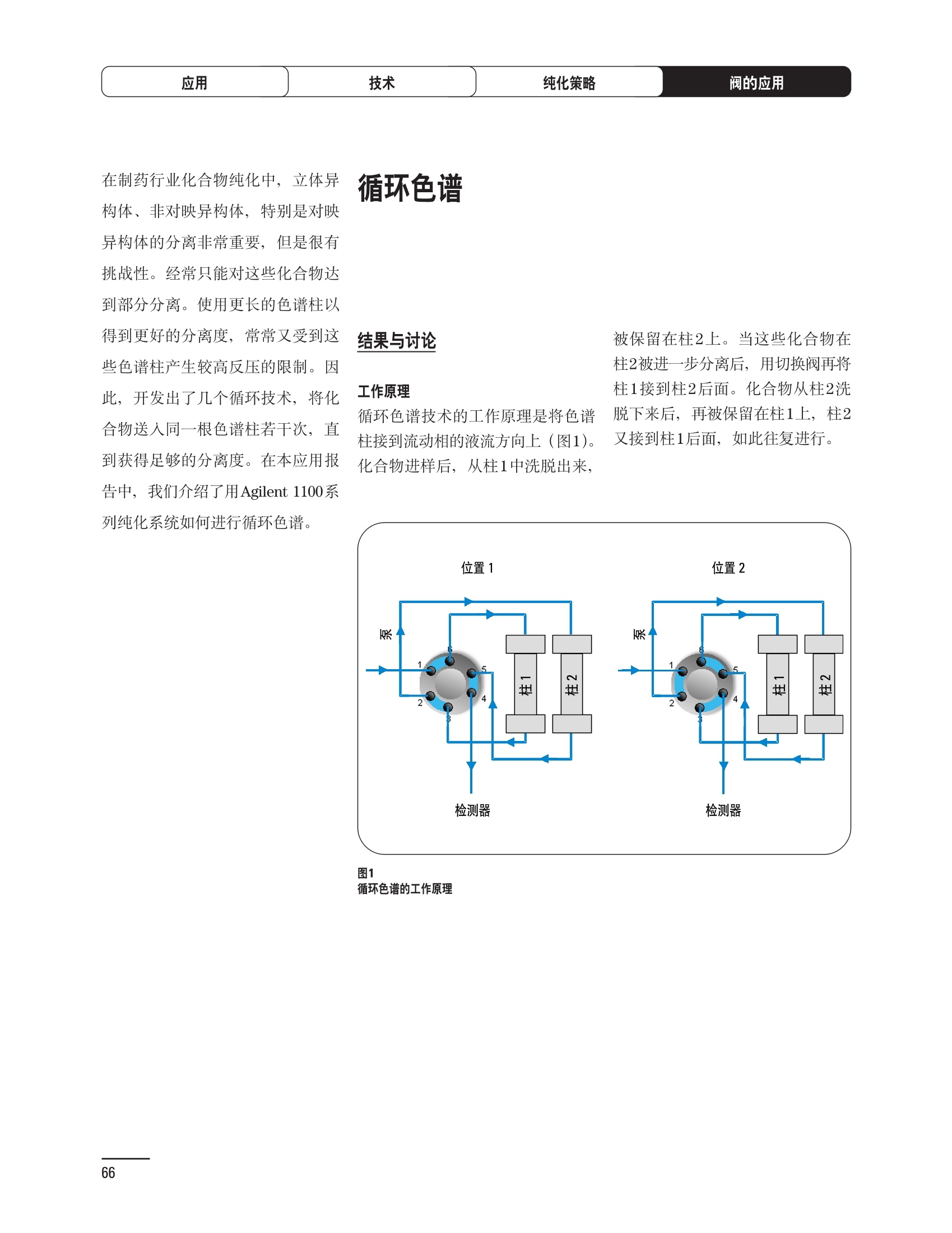
制备液相色谱解决方案 ● 应用概论 Agilent Technologies 目录 应用-相关报告... 2-15 用制备液相色谱分离杂质. 2 临床前研究尿样中放射性标记药物代谢物的分离与纯化. 4 人尿样中候选药物代谢产物的分离与纯化.... .6 对映异构体的纯化.... 8 对化合物库进行质谱引导的馏分收集. 天然产物的纯化.. 蛋白质的纯化与鉴定.. 14 技术-相关报告... .16-51 用Agilent 6110/6120四极杆LC/MS系统进行质谱引导的馏分收集 质谱引导的纯化系统的最佳配置. 在高流速下进行质谱引导的馏分收集. .20 质谱引导的馏分收集的优化.... .22 用安捷伦双定量环自动进样器进行性能优化... .24 等度纯化实验中的多次进样.... 分析与制备的组合系统. 进样泵系统... 回收收集. 在高流速下进行质谱引导的的馏分收集 质谱引导的馏分收集的优化.. .36 常规馏分收集的优化.. .38 优化系统以获得最大回收率.... 基于第三方检测器的色谱谱进行馏分收集.. 方法的放大和放大计算.. 流速从1到100 mL/min的制备泵性能.. 远程数据浏览提高实验室效率. 纯化策略. .50-65 根据预制备性分析建立优化的制备方法 针对药物化学类别制定化合物纯化策略 在配备质谱检测器的系统上进行化合物纯化... 对未达到基线分离的化合物进行纯化.. 基于色谱峰的馏分收集中正负斜率的应用... .58 自动馏分收集后再分析中可能出现的误差.. 高浓度样品的进样.. 高效率的轻松操作. 纯化系统阀的应用. ..66-71 循环色谱. ..66 制备液相色谱中的交替柱再生. 回收收集和基于时间的馏分收集.. 本书集中介绍了在Agilent 1200和1100系统纯化平台上所进行的各种研究和应用实例。探讨了其他专业人士的各种经验,内容涉及:馏分收集与再分析、基于色谱峰和质量数的馏分收集,以及纯化系统的优化。 本书摘要介绍了一些安捷伦制备液相色谱的应用报告。这些在Agilent 1100系统上进行的纯化应用,都已经在Agilent 1200系列液相色谱系统上进行了验证,后者与Agilent 1100系统相比性能相当或更好。本书中的应用报告以概要形式介绍,如果需要深入阅读全文,可从安捷伦网站上下载,网址: www.agilent.com/chem/purification。 ·选择泵、自动进样器、馏分收集器、检测器、色谱柱和制备型流通池,对每个应用进行优化,以获得最大的回收率和最高纯度 。可扩展的选择,让您能够根据样品量(从微克到克级)制定纯化方法 ·基于时间、色谱峰和/或质量数的馏分收集-独有的组合 ·使用简便的软件让非专业用户能够进行便利的无人执守操作 。为您的所有设备、备件和技术支持提供单一厂商解决方案 ( 1200系列纯平平台为分析型和制备型应用提供了两种基本平台: ) ( ·1200系列纯化系统AS(分析型),流速低于5 ml/min ) ·1200系列纯化系统PS(制备型),流速最高至100 ml/min 1200系列纯化系统AS 1200 系纯纯化系统PS 杂质分析是要研究一组分析活性化合物,目的是对药物中的有机和无机杂质进行检测、鉴别、结构解析和定量测定。在这一过程中的首要任务,就是对所有杂质进行检测。即使采用最精密的质谱仪,也不可能对所有化合物进行完全的结构解析。需要对这些化合物进行分离和纯化,然后再用1H-和13C-NMR进行鉴定。本应用报告报导了一对不能用MS鉴定的杂质的分离和纯化。 结果与讨论 图1中的杂质A和D可以用离子阱和TOF质谱进行鉴定,而杂质B和C则不能被完全鉴定,因此要用HPLC分离,进一步做结构分析。 方法优化 MS实验中所用的2 pm以下填料固定相也可以用5-um粒度填料的制备柱代替,只需要对方法进行优化,以调整杂质B和C的分离度,并缩短运行时间。含杂质B和C的样品分离结果见图1。 0制制备液相色谱分离杂质 图1 杂质C和B分离方法的优化 纯化参数 采用化学工作站的馏分预览优化馏分收集参数。图2中是制备分离的色谱图,调节馏分收集参数,如阈值、正斜率、负斜率和上限阈值,直到得到合格的馏分收集性能。在这个例子中,在一定时间窗口内(9-14分钟)基于阈值进行馏分收集就能得到最好结果。图3显示了采用馏分预览所优化的参数,实际进行馏分收集的结果。汇集10次连续分离纯化后的馏分,分析结果见图4。收集到了足够纯度的杂质B和C后,经NMR分析(数据未显示)得到的结构解析结果见图4。 图3 结论 馏分收集结果 本应用报告报道了两种杂质的分离和纯化,首先在Agilent 1200系列高分离度快速液相色谱系统上建立了高分离度分析方法。因为固定相可以用2 um填料,也可以用标准的5 pm粒制备柱,所以能够将分离方法从标准的Agilent 1200系列完整过渡到Agilent 1200系列制备系统上。因此,经过方法优化和上样实验后可以直接放大方法,不需要在制备柱上再进行方法优化。收集到的组分具有很高纯度和回收率,使杂质结构解析能够轻松完成。 图4 药物合成产物中杂质的鉴定 用Agilent 1200系列LC/MS系统进行杂质分析-第2部分:用制备液相色谱分离杂质,发行号5989-5618EN。 应用 技术 纯化策略 阀的应用 鉴定新药候选药物代谢产物是药物开发过程的一项基本工作。在早期药物研究与优化中发挥着重要作用,由此找到具有更好药代动力学和预计特性的候选药物。药物开发后期,鉴定实验动物和再后来鉴定人体的药物代谢产物,是法规要求的安全性实验。在药物开发中,药物代谢研究通常是用放射性标记的候选药物完成的,所以很容易用放射化学检测鉴定相关代谢产物。代谢物通常是以低浓度存在于非常复杂的基质中,如尿、胆汁、血浆,要用核磁共振(NMR)波谱等技术对代谢物进行准确鉴定,就必须先对其进行分离纯化。 狗尿样品进样 图2是10 mL狗尿样品的放射化学检测器和DAD检测图谱, RD信号中很容易鉴别出放射标记候选药物及其代谢产物。分离代谢产物,可以用RD监测按时间进行馏分收集,也可以根据RD信号按色谱峰进行收集。 前体化合物 ~1ug/mL水溶液 进样10mL 根据RD信号按色谱峰进行馏分收集 为了能够根据来自RD的信号触发馏分收集,将信号输出连接到通用接口盒(UIB)上,后者与Agilent 1100系列组件的CAN网络连接。为了确保没有代谢物丢失,甚至在馏分收集器触发失败时也不发生丢失,在系统上配置了一台回收收集器。基于色谱峰的馏分收集结果见图2。 结论 本应用报告报道了一套纯纯系统,并将这一优化的系统用于分离纯化来自典型DMPK的放射性标记物。这一配置的最重要部分是可以大体积进样的进样泵系统,, 1以及用放射化学检测器鉴定含代谢产物的馏分。另外,对GlaxoSmithKline, Ware,UK.DMPK小组代谢研究的实际样品的分析表明了收集策略的正确性。 图2 根据RD信号按色谱峰进行馏分收集 应用 技术 纯化策略 阀的应用 鉴定新药候选药物代谢产物是药物开发过程中的一项基本工作。在早期药物发现与优化中发挥着重要作用,由此找到具有更好药代动力学和累积特性的候选药物。药物开发后期,鉴定实验动物和再后来鉴定人体的药物代谢产物,是法规要求的安全性实验。人体实际代谢的研究通常要在临床研究中定量服用放射性标记药物。志愿者服用候选新药后,收集人体代谢初步数据,是I期临床研究的内容。这时候需要将代谢产物从大体积的生物基质中分离出来,这个例子中的基质是人尿,然后用核磁共振(NMR)波谱学等技术进行结构鉴定。 基于时间的馏分收集用于人尿样品 图2显示了50 mL人尿样品用进样泵系统进样后,按时间进行馏分收集的结果,每0.5分钟将馏分收集到12mL试管中。所有收集的馏分。可继续使用NMR或其其LC/MS技术进行进一步结构分析。 图1 鉴定含药物及其代谢物的馏分 候选药物及其某些已知质量数的代谢产物,可以用MSD鉴定。含5种化合物的馏分很容易地得到了鉴定,因为馏分中每个标记的信号都可以用DAD和MSD延迟时间排列。 结论 本应用报告报道用进样泵系统进样了50 mL人尿, 以及在Agilent 1100系列纯化系统上按时间对候选药物及其代谢物进行了馏分收集。用MSD监测馏分收集结果,用提取离子流色谱图找到了4个代谢产物和一个前体化合物。很容易地鉴别出含有这些化合物的馏分,为进一步分析提供了半纯化的馏分。 图2 对人尿样品基于时间的馏分收集 应用 技术 纯化策略 阀的应用 药物的立体异构体组成是目前药物开发中的一个关键问题。药品申报需要纯的药物异构体,在筛选阶段需评价两种异构体在疗效、毒性或药代动力学方面的差异。因为,实际上在早期开发中进行相关实验只需要有少量的有效药物,所以制备高效液相色谱是从消旋体中分离纯异构体的适当工具。在本应用报告中报道了克级呼吸系统候选药物外消旋体的分离。分离在Agilent 1100纯化系统上进行。 分析方法 在Chiralpak AD-H5 pm柱(4.6x150mm)上建立了分离对映异构体E1和E2的方法。该方法在纯化阶段后期还用于控制馏分的纯度和馏分中的对映异构体过量。实验测定了能加载到分析柱上的最大样品量。 合作者Volkmar Korner,Boehringer Ingelheim, Germany 制备方法 根据在分析柱上进行的过载实验结果,在制备柱上进行了放大。为了增加样品制备量,将样品浓度增大到5050和200 mg/mL (乙醇溶液),进样体积保持在900 pL。分离结果见图1。 合并馏分的分析 用分析方法中所描述的方法对合并的馏分进行了分析。两个异构体得到了良好分离,对映体高度过量,见图2。 结论 本应用报告报道了用Agilent 1100纯化系统对德国Boehringer Ingelheimin Biberach, 研发的呼吸系统候选药物中的两个对映异构体进行分离。该方法在分析柱上建立,进行过载实验后放大到制备柱上。调整制备方法后,完成了克级纯化,将收集到的馏分合并后再分析,测定对映异构体过量。 图2 合并馏分的再分析 化合物库含有需要筛查生物活性的一系列结构类似的化合物。虽然,组合化学和常规合成化学相比,简化了合成过程,但仍然需要从其杂质和反应副产物中对化合物进行纯化。 仪器 仪器包含了两个流路。主要流路是从二元泵到自动进样器、柱温箱、二极管阵列检测器和馏分收集器之前的主动分流器。由于质谱检测器(MSD)是有损检测器,主要流路的流速过高,不能直接进入电喷雾离子源,所以要用一台等度泵保持一个辅助液流。这一辅助液流来自等度泵,流人MSD之前的主动分流器。为了使质谱检测更容易,主动分流器将主液流中的一部分汇入辅助液,随后者进入MSD。 结果与讨论 样品制备 为了说明用Agilent 1100系列纯化平台进行质谱引导的馏分收集结果,我们建立了包含17种相关化合物的库,这些化合物涵盖了吩噻嗪的所有衍生物(图1)。 图1 吩噻嗪衍生物的结构 质谱引导的馏分收集 在软件中设定了17个混合物中每个要收集馏分的目标质量数。根据单电荷正离子触发馏分收集,见图2。 用预先设定的质量数进行馏分收集,与常规的非特异性检测器相比,有如下优点: ·将这项技术用于每一次分离,只收集感兴趣的化合物 ·不用在色谱分离中从-——一系列收集的馏分中挑选目标化合物 ·安捷伦专利的馏分收集延迟校正确保了可靠的样品回收 总之, Agilent 1100系列为质谱引导的馏分收集提供了节省时间和资源的纯化手段。 图2. 12个选定TIC色谱图。图中的标记显示了馏分收集的开始和结束 这是一个用Agilent 1100系列纯化系统AS和PS对红三叶中刺芒柄花素和其它植物性雌激素进行分析型和制备型分离的例子。 天然产物的纯化 结果与讨论 体积过载实验/上样量实验 因为红三叶提取物的浓度是一定的,所以不可能进行浓度过载,只能用体积过载分离目标化合物。粗提物进样50pL对于分析规模的纯化仍有足够的分离度。 植物性雌激素的分析型分离 从复杂天然提取物中分离各种化合物的通用方法,是按时间段收集馏分。由于过载实验得到了良好的分离,所以对红三叶提取物采用了基于色谱峰的馏分收集。 更大量的分离 为了获得更多的植物雌激素物质,将几次分离得到的馏分汇集到一起。即,从一个样品瓶中重复进样,将每次的馏分都收集到同一个试管中。 9次50-pL进样共进样450 pL, 将馏分自动合并。对该馏分的再分析显示结果良好,说明仪器和软件都具有卓越性能(图1)。 另一个纯化更多物质的可行方法是放大到更大的色谱柱上。根据分析柱的过载实验,进行放大计算,次进样450 uL。在9.4x150mm柱上进行,流速7 mL/min。因为Agilent1100系列多孔板自动进样器AS最高流速只能到5mL/min,所以将纯化转移到纯化系统PS上进行。 植物性雌激素的制备型分离 为了纯化更大量的样品,将方法进一步放大到21.2x150 mm柱上。在这种规模的色谱柱上, -次进样可以注入2300pL样品。色谱图见图2。馏分再分析显示,放大没有造成馏分纯度方面的任何损失。 分析型放大方法可用于在Agilent1100系列纯化系统AS上分离复杂植物粗提物中的化合物。在这个方法的基础上,进行了按色谱峰收集馏分的制备型分离。 。为了获得更多的植物雌激素物质,使用了Agilent 1100系列纯化系统的馏分合并功能 ·通过对馏分的再分析测定了所得化合物的纯度 ·根据分析结果将方法放大 。为了在一次运行中得到更大量的目标化合物,在Agilent 1100系列纯化系统PS上用两种不同的色谱柱重复了纯化方法 制备规模的结果与在分析型系统上得到的结果相当。 图1 合并馏分的再分析 图2 21.2-mm制备柱上的馏分 ( I solation of Formononetin and Other P h ytoestrogens from Red Clover with the Agilent 1100 Series Purification Sy s tem" (用Agilent 1100系列纯化系统分离红三叶中的刺芒柄花素和其它植物雌激素),发行号5988-5747EN。 ) 蛋白质的产率和纯度是衡量纯化是否成功的重要指标。除有效的色谱分离外,配置良好、可靠的仪器也能影响纯化的成功。制备型反相(RP)液相色谱由于具有高分离能力,经常被用做多肽和小分子亲水蛋白纯化流程的最后一步。虽然大家都知道反相液相色谱所用的溶剂条件会使蛋白结构变性,但也能通过调节适当的条件再使其复性,特别是小分子蛋白。Agilent 1100系列纯化系统是设计良好、合适的反相色谱的系统。 结果与讨论 在Agilent 2100生物分析仪上只用了4pL预纯化的样品,即完成了第一个分析步骤,确定56kDa感兴趣的蛋白质是否存在,有多大的量。图1显示了分析的电泳图谱和拟凝胶图像。电泳图中28秒处的峰相对于58.6kDa蛋白,占总蛋白浓度的20%,在拟凝胶图像中也看到了同样的结果。这清楚地表明样品中存在目标蛋白。 剩下的原料将用反相液相色谱进行纯化。图2显示了用Agilent 1100纯化系统对预纯化样品反相液相色谱纯化的色谱图。竖线指示了馏分收集的起始和终止,水平线代表触发馏分收集的阈值。另外,馏分收集器上的相应试管对应于各个馏分。 为了鉴定出含56 kDa目标蛋白的馏分,将三个馏分全部冻干,溶解在PBS中, 用Agilent 2100生物分析仪分析。图3a显示了起始原料和三个馏分的电泳图。相应的拟凝胶图像见图3b。电泳和相应的拟凝胶图像数据清楚地表明,56kDa目标蛋白在馏分2中得到纯化。图4a显示了对馏分2的反相液相色谱再分析。由于使用了精确的延迟体积校正,再分析的色谱图显示在11.5分钟左右有一个单一的对称峰。虽然结果提示是纯的蛋白,但馏分2的电泳图(图4b)仍显示在20kDa附近有一个小杂质。用2100生物分析仪软 件测定的蛋白纯度表明,456 pg/mL中该蛋白相对浓度为76%。这个结果很好理解,我们知道Agilent 2100生物分析仪分离蛋白的方法是与反相液相色谱互不相同的。已知反相液相色谱根据疏水性不同分离蛋白分子,而2100生物分析仪则检测蛋白尺寸之间的差异。由于这个原因,图4b中所见的杂质比较明显。在反相液相色谱色谱图中,该峰包含了两个不同的共洗脱蛋白。要得到纯蛋白,还需要用互补方法进一步纯化,如可以用离子交换色谱或凝胶过滤。 结论 如果样品纯度是至关重要的问题,那就不能只用一种分析方法测定。基于这个理由,安捷伦提供了解决方案-1100系列纯化系统和2100生物分析仪,二者结合,确保进行有效而可靠的蛋白质纯化和鉴定。 图1 图2 在Agilent 2100生物分析仪上用Protein 200 Plus试剂盒分析预纯化的蛋白样品。显示了电电图谱和拟凝胶图像。数字为分子量,以kDa表示。56 kDa感兴趣蛋白的迁移时间为28s, 对应于58.6 kDa 预纯化样品的纯化色谱图。竖线分别表示馏分收集的起始和终止。水平线代表280 nm UV信号的峰触发阈值。另外,数字表示在馏分收集器上所收集馏分的位置 图3a/b 在Agilent 2100生物分析仪上用 Protein 200 Plus试剂盒分析纯七系统得到的三个馏分。显示了电泳图(A)和拟凝胶胶像(B)。数字表示以kDa为单位的分分量。56kDa感兴趣蛋白在馏分2中 图4a/b 馏分2的色谱图(A)和电泳图(B)。虽然反相液相色谱表明是纯蛋白,但电泳图仍显示在20 kDa左右有一个小杂质 ( P rotein Purification and Characterization Using the Agilent 1100 Series Purification System and the 2 100 Bioanalyzer (用Agilent1100系列纯化系统和2100生物分析仪进行蛋白质纯化和表征),发行号5988-8630EN。 ) 应用 技术 纯化策略 阀的应用 Agilent 1200系列纯化系统是药物高通量合成小分子和大分子化合物库纯化的通用工具,能够得到高纯度馏分,获得目标化合物的高回收率。Agilent 6110/6120系列MSD是新一代单级四极杆仪器,可以进行简便而可靠的质谱引导的馏分收集,也可以完成更复杂的纯化工作,如组合离子源或使用正/负极切换等。 结果与讨论 用组合离子源进行质谱引导的馏分收集操作安捷伦组合离子源需要Agilent6120 MSD。可以同时进行电喷雾和大气压化学电离。化合物在ESI或APCI中电离后,将产生离子,并触发馏分收集。质谱引导的馏分收集结果见图1。 用组合离子源和正负极切换进行质谱引导 的馏分收集用Agilent 6120 MSD, 还能在一次运行中进行正负极切换。如果目标化合物以正离子或负离子模式在ESI或APCI中电离,都能产生离子。一般在方法中都指定[M+H]+和[M-H]-等加合物,以触发馏分收集。质谱引导的馏分收集实验结果见图2。 结 本应用报告报道了用不同电离技术,如ESI和APCI, 以正电离和负电离模式,用Agilent 6110/6120 MSD进行质谱引导的馏分收集。可以用Agilent 6110 MSD ESI或APCI源,正或负离子模式,质谱引导的进行简便可靠的馏分收集。如果想要在一次运行中使用组合离子源,同时进行ESI和APCI, 就需要使用Agilent 6120 MSD。 图2 组合离子源、正/负电离模式,质谱引导的馏分收集结果 "High Performance Mass-based Fraction Collection with the Agilent 6110/6120 Quadrupole LC/MS System Equippedwith Multimode Source”(用配备了组合离子源的Agilent 6110/6120四极杆LC/MS系统进行高效的质谱引导的馏分收集),发行号5989-5673EN。 质谱引导的纯化系统的最佳配置 因为合成化学家已经知道了药物或高通量合成化合物的分子量,所以质谱引导的馏分收集的制备型液相色谱是经常选择的纯化方法。质谱引导的收集馏分的一个重要部件是分流器,它对所收集馏分的纯度有很大影响。本应用报告讨论了分流器的设计和流速分流器的正确配置。 合作者Martin Fuhr, JudithStefifens, Griinenthal GmbH,Germany 结果与讨论 配置1: 分流器的设计 分流器之前的UV检测器 市场上最多的流速分流器是被动分流器(图1a)。分流是通过使用各种长度和内径的管路实现的,这将造成不同的反压。安捷伦的主动分流器(图1b)有独特的工作原理:用一个切换阀将主液流中一定体积的流动相主动转移到辅助流中。分流比由阀的切换频率决定。 这种配置(图2)只能使用主动分流器,因为被动分流器会给UV检测器的流通池带来太高的反压。UV引导、质谱引导和合并馏分收集的纯度结果见表1。 图1 配置2: UV检测器在分流器和MSD之间 在配置2中分别用主动分流器和被动分流器进行了实验。结果见表2。用被动分流器得到的结果总是比主动分流器的略低。而且比用配置1的纯度低,特别是采用质谱引导的进行馏分收集时。 配置3: 至UV和质谱检测器的第二分流器 在这个配置中,来自主动或被动分流器的液流,用一个三通再次分流到UV和质谱检测器。通过由检测器反压得到的分流比没有测定。在配置3中用主动分流器和被动分流器进行了实验。结果见表3。 图2 配置1:UV检测器在分流器之前 分流器 收集 纯度尼莫地平 主动 MS 99.4% 92.1% UV 与MS 98.8% 被动 W 97.3% 81.0% UV 与 MS 93.0% 表2 配置2的纯度结果 结论 能得到最好纯度结果的配置,特别是在质谱引导的馏分收集中,是将分流器直接放置在UV检测器之后。但是,这种配置只能用主动分流设置,因为被动分流给UV流通池造成的反压太高。图3总结了纯度实验的结果。 分流器 收集 纯度尼莫地平 主动 Ws 98.3% 89.2% UV与MS 95.2% 被动 MS 98.2% 83.1% UV与 MS 94.2% 表3 配置3的纯度结果 图3 纯化实验的结果 通常用自动化的制备型LCMS系统对药物研发中的组合化学化合物库进行纯化。30到50 mg化合物组成的混合物在20 mm内径的色谱柱上分离,流速20到35 mL/min, 采用质谱引导的馏分收集。到目前为止,需要几克起始原料进行的合成化合物库的纯化,还要靠经典的纯化方法,如结晶或硅胶上的手动flash色谱。在本应用报告中,我们报道了用Agilent1100系列纯化系统,用质谱引导的收集馏分模式,在100mL/min流速下,在50 mm内径柱上,-一次运行就纯化了4克粗产品。 100mL/min流速下质引引导的收集馏分的系统配置 要得到100 mL/min高流速,用0.7-mm i.d.毛细管将制备泵连到双定量环自动进样器,把自动进样器连接到色谱柱上。用0.06mm光程长度制备流通池配备的0.8-mm i.d毛细管连接色谱柱和UV检测器。在主动分流器和馏分收集器之间安 装一个10-mL毛细管,因为在MSD和馏分收集器之间会有5秒钟的延迟时间。在双定量环自动进样器上安装一个10-mL样品定量环,用于大体积进样。 ( 分析型馏分收集器用0.8-mmi.d.制 备管路工具包(G1364-68711)改造, 并配备高流速针头(G 1 364-87202)和 40位漏斗盘(G1364-84512),用于收 集大体积馏分。 ) 克级纯化 将测试混合物A(单个化合物1333 mg溶于12000 pL DMSO中)注入填充了安捷伦制备C18填料的50x200 mm柱(制备量约4克)。按照触发质量数441收集尼莫地平,因为该化合物有较强的加钠峰(418加上加合物[M+Na]+即为触发质量数441),见图1。 对收集的馏分进行再分析显示尼莫地平纯度为96.2%(根据UV信号峰面积计算),如图2所示。 结论 图1 质谱引导的馏分收集的结果 本应用报告报道了用50-mm内径柱以100 mL/min流速,质谱引导的对少量化合物的纯化。即使因为系统的某些小改动使色谱峰没有基线分离,化合物也能实现高纯度分离。即使使用光程非常短的制备型流通池,[UV信号不再能分开的样品也能得到纯化。 图2 收集馏分的再分析 ( Mass-based Fraction Collection at High Flow Rates for t he Purification of Compounds in the Lower Gram Scale" (以高流速质谱引导的馏分收集对低克级化合物进行纯化),发行号5989-2469EN。 ) 在现代纯化系统上进行馏分收集,可以根据保留时间窗口、检测器信号,也可以根据来自MSD的目标化合物质量。MSD具有选择性更高的优点,从而使收集的馏分数量更少,馏分中肯定含有目标化合物。 在本应用报告中,比较了基于色谱峰和质谱引导的馏分收集触发技术的目标产物的纯度和回收率,阐述了各种触发技术的优缺点。 结果与讨论 质谱引导的馏分收集 质谱引导的馏分收集系统必须包含一个分流器,将来自色谱柱的液体分流到馏分收集器和MSD。根据设计,分流器都或多或少地导致色谱峰在MSD内变宽。另一个使峰变宽的原因是MSD本身。因为MSD是-种浓度依赖型检测器,是为最高灵敏度分析而设计的,当制备样品进入系统时,如果浓度很高,将出现过载。 根据UV和MSD信号的逻辑量“与”进行馏分收集 在纯度和回收率方面具有最好的纯化结果并保持了MSD的选择性的纯化,是将UV和MSD信号按逻辑“与”的关系来进行收集,如图1所示。一个馏分只有同时满足UV及MSD的触发指标,才能进行馏分收集,即,如果有UV信号,只要指定的目标质量数不存在,就不会收集峰。 根据两个化合物的分离度,收集一个或两个馏分。结果表明,大部分目标化合物在第一个馏分中以高纯度存在,第二个馏分中收集到了一些其它化合物和杂质。对馏分1和馏分2的再分析(图2A和2B)显示,馏分1中含有9.76 mg 尼莫地平,相当于回收率92.5%,纯度95.8%。馏分2中含有另外0.01mg的尼莫地平,尼莫地平的整体回收率为92.6%。 结论 表1中总结了不同触发模式的优缺点。基于色谱峰的馏分收集,如根据UV检测器信号,如果将检测器设置在柱后(不带分流器),能得到纯度和回收率最好的纯化结果。使用MSD提高了系统的选择性,但降低了所收集馏分的纯度,这是由于分流器和MSD本身使峰变宽造成的。最好的结果是将MSD的选择性和UV检测器所得的良好峰形以逻辑“与”结合起来得到的。但只能使用不增加系统反压的分流器,才可能将UV检测器直接安装在分离柱后,而不是分流器之后。 图2 A)馏分1的再分析,B)馏分2的再分析 触发模式的比较 ( 0ptimizing Mass-based Fraction Collection for Highest Purity using the Boolean Logical-AND-Combination with theUV Signal ” (用Boolean逻辑 “与”结合UV信号,优 化 质谱引导的最高纯度的馏分收集),发行号5988-2014EN。 ) 应用 技术 纯化策略 阀的应用 双定量环自动进样器(DLA)的进样原理与其它安捷伦1100系列自动进样器不同。在DLA中样品先吸入缓冲定量环,再用低压流速计转入样品定量环中。然后样品定量环再切换到高压流路。这种称为固定定量 环进样的原理,有两个操作模式: 部分充满,这时样品体积比样品定量环的体积小,进样不会损失任何样品 。定量环完全充满,精确度高,但损失了吸入的很大样品体积 部分定量环充满是制备操作选择的方法,而完全充满适用于分析任务。 用安捷伦双定量环自动进样器进行性能优化 工作原理 完全和部分充满 DLA的固定定量环概念可以有两种操作模式-定量环完全充满和部分充满。当需要高精密度进样时,必须使用完全充满,即必须从样品中吸取大于定量环体积的样品。如果进样精密度不是很重要,而重要的是不要浪费样品,则应该选择部分充满。从样品容器中吸取再注入定量环的样品体积比样品定量环的体积小(图1)。完全和部分定量环充满的优缺点总结如下: ·完全定量环充满 -定量环必须过量充满(进样量是定量环的3到5倍),大部分样品流入废液瓶 -高精密度→分析任务的理想选择 ·部分定量环充满 -所有样品都进样到柱上 -精密度较低(与样品定量环大小、样品体积和吸取速度等因素有关) →制备任务的理想选择 性能 1.完全定量环充满 定量环完全充满是要满足分析型液相色谱最高进样精密度的要求而选择的方法。当溢出因子最小为1时,不会得到准确而精密的结果。图2显示了从1到5不同溢出因子所得到的峰面积(50-pL定量环)。可以看出,溢出因子从3到5,峰面积明显增大。也就是说,溢出因子低于3时,样品定量环没有完全充满。 2.部分定量环充满 制备HPLC的目的是把从样品容积中吸取的全部样品,全部进样到色谱柱上,而进样精密度并不重要。 图1 定量环部分和完全充满 样品定量环充满因子 图3显示了用同样的样品定量环(500 pL),几次不同进样体积的进样结果。在定量环充满50%以前,峰面积呈线性增加(用点状线表示)。这说明,为了最大限度地减少样品损失,最大进样体积不应高于定量环体积的50%。 吸取和推出的速度 吸取和推出的速度对峰面积精密度没有影响,但影响回收率,如图4所示。 结论 要用Agilent 1100系列双定量环自动进样器取得最佳性能,,需牢记以下几点: 定量环完全充满 ·要得到最高的面积精密度和最好的分析结果,必须使用定量环完全充满法。 。要得到最好的进样准确度和精密度,溢出因子必须在3到5之间。 定量环部分充满 制备工作必须使用定量环部分充满,让从样品容器中吸取的样品全部注入色谱柱。 ·要得到最好进样准确度和最高的样品回收率,样品定量环不应该充满,不要超过定量环体积的50%。 图2 不同溢出因子的峰面积 图3 定量环部分充满时峰面积的线性变化 图4 定量环部分充满的性能-吸取/推出速度的影响 制备液相色谱是目前药物开发中纯化化合物所选择的方法。大量粗品用通用方法得到纯化,采用短和快速梯度在约10分钟之内完成。虽然大部分化合物可以通过这些方法得到分离,但分离和纯化立体异构体或非对映异构体需要更长的运行时间,采用等度条件或非常平缓的梯度。在本应用报告中,我们报道了如何在一次运行中多次进样以提高样品产量,节省了宝贵的时间和流动相。 在第一个异构体洗脱约25分钟之后,5次进样的总运行时间为70分钟,样品中没有其它杂质时,可以再次 流动相消耗1470 mL。与5次单独进进样。这一间隔由第一个峰起点到 样相比(175分钟,3675mL),相当第二个峰终点之间的时间差决定, 于节省了60%的时间和流动相。大约7分钟。5次连续进样的结果见图1。 进样器程序 多次进样的进样器程序有10行,包括将样品推出样品定量环后清洗针头表面和冲洗针头、针座、针座毛细管及阀。根据一次运行中的进样次数,对这10行命令进行复制。 局限和限制 ·必须用流动相溶解样品 ·在Use Loop项下Set up Injector窗口必须设置多次进样所用的样品定量环 进样器程序中的行数限制在60步以内 。要减少进样器程序的行数,可以 使用REPEAT/END REPEAT命令 分离纯化异构体和对映异构体时,在一次等度运行中连续进样,可以节省宝贵的时间和流动相。使用Agilent 1100系列纯化系统,可通过设置进样器程序轻松进行多次进样。 ( P erforming Multiple Injections in an I s ocratic Purification Experiment using the Agilent 1100 Series PurificationSystem n” (等度纯化实验中用Agilent 1100系列纯化系统进行多次进样),发行 号 5989-1651EN。 ) 将分析与制备工作结合在一起的系统始终要兼顾硬件的性能,如毛细管的内径。本应用报告阐明如何用最好的配置设置这样的系统。 结果与讨论 基本概念 模式的选择 通过选择种方法确定使用分析还是制备模式。至少有一个分析方法和一个制备方法。用选择阀在分析和制备模式之间切换,,引导来自四元泵的流动相,或来自制备泵的流动相,再通过双定量环自动进样器。在制备模式中,四元泵作为辅助泵。 主动分流器在制备操作中,分流器打开,用四元泵提供辅助液流。在分析操作中,分流器关闭,来自四元泵的液体流经双定量环自动进样器进入分析柱、二极管阵列检测器,辅助流从安捷伦主动分流器到MSD。 图1 双定量环自动进样器 双定量环自动进样器包含两个进样定量环。制备模式配置的是2000-pL定量环,使用部分定量环充满技术。分析模式使用的是50-uL定量环,使用部分或完全定量环充满方式,取决于分析样品的浓度。 应用实例 下列样品应用在按前面所述的设置和配置的系统上进行。制备分析中,将UV引导(仅根据斜率,正斜率150 mAU/s)和质谱引导(仅根据阈值,150000计数信号)的馏分收集设定为逻辑“与”的关系,即只有二者的条件都满足才进行收集。结果如图2所示。 图2 应用 技术 纯化策略 阀的应用 由于样品化合物的溶解度不同,样品进样体积在从几微升到几升之间变化。5 mL以下的小体积样品可以直接用自动进样器进样,而更大的样品体积通常要用进样泵进样。如果使用高压进样泵,样品可以被直接泵人色谱柱。本应用报告报道了将Agilent 1100系列等度泵作为高压进样泵的纯化系统。 结果与讨论 系统配置 该系统的配置见图1。用一个2-位/六通阀在提供梯度洗脱的制备泵和进样泵之间切换。整个纯化运行包括三个步骤: 合作者Graham Foster, Richard0Hanlon, GlasoSmithKline, UK 1.最初,进样阀位于1位,此时来自梯度泵的流动相进入色谱柱,进样泵的液流流入废液瓶 2.开始运行后,进样阀切换到2位,此时来自进样泵的液体进入色谱柱,进样泵把样品转入色谱柱 3.样品加载到色谱柱上之后,进样阀切换回1位,开始进行梯度分离。在对色谱柱进行梯度洗脱的同时,用适当溶剂清洗进样泵和进样阀 样品进样系统 样品进样系统包含进样泵和一个12位/13-通阀,阀的出口与泵连接。几个容器分别装有梯度起始溶剂(溶剂A)、样品和清洗溶剂,按下列次序连接在入口位置(图2): ·1位:起始位置,溶剂A(梯度起始组成) ( ● ·2位 : 样品 ) ·3位:溶剂A冲洗样品,通过阀和进样泵,完全注入柱上 4位:清洗溶剂冲洗进样阀和进样泵 ·5位:溶剂A将清洗溶剂从进样泵和进样阀中置换出来 泵和阀切换 通过执行“Sample injection system”项下编辑的工作流程,为梯度泵、进样泵、进样阀和样品阀设置时间表进行。 系统的范围和局限 ·系统用安捷伦化学工作站操作 ·根据化学工作站顺序设置基于时间或色谱峰进行馏分收集。只有设置单次运行(Run Method task)时,才能进行质谱引导的馏分收集 ·进样循环由梯度泵、进样泵、进样阀和样品阀时间表控制 ·不能使用延迟体积校正。等度泵的最大流速为10 mL/min ·对于高度浓缩的样品,建议从等度泵的放空阀中取下PTFE过滤头(操作步骤见等度泵手册,部件号G1310-90003) 图2样品进样系统 结论 在本应用报告中,我们报道了配有一个等度泵和一个简单的阀解决方案的进样系统的Agilent 1100系列纯化系统的设置和配置。描述了如何用泵和阀时间表设定方法。 应用 技术 纯化策略 阀的应用 有时无论采用什么样的触发机制,都希望不仅收集感兴趣的化合物,而且还要将样品中的所有组分都收集到称为回收区的特定容器中(图1)。回收溶液可以干燥后进行再回溶分析,以回收在第一次纯化中丢失的感兴趣样品组分。 讨论 回收收集-为什么? 回收溶液可以用来回收样品中第一次纯化未触发的任何组分。同时这也是一项安全回收措施,例如,如果纯化系统没有收集到预期目标化合物时。 回收收集-如何操作? 必须从馏分收集器的废液管路收集回收溶液。如果系统中配置了不止一个馏分收集器,则必须在回收收集之前将废液管路合并。 用Agilent 1100系列纯化系统进行回收收集用Agilent 1100系列纯化系统进行回 收收集必须用三种方式进行: ·在馏分收集器废液管路中安装一个12位/13通阀-有12个回收区-有通过软件追踪的基本回收区 ·第三方馏分收集器和BCD板 -回收区的数量取决于馏分收集器 -没有软件追踪的回收区 ·Agilent 1100系列馏分收集器-最多120个带漏斗盘的回收区-可追踪的完全回收区 112立/13通阀 馏分收集器的废液管线连接到阀的 入口位置,12个出口位置分别连接 回收容器。每次进样时,阀都会自 动切换到下一个位置。 第三方馏分收集器和BCD板 安装在自动进样器中的BCD板,为自动进样器的瓶号和4个外接点提供BCD输出。用通用电缆连接BCD输出到外接点到外接设备。这4个外接点可以用化学工作站的进样器程序打开和关闭,让第三方馏分收集器在一次进样后切换到下一个位置,或切换分流阀。 Agilent 1100系列馏分收集器 用化学工作站A.10.01或更高版本,可以在系统中配置一个另外的Agilent 1100系列馏分收集器进行回收收集。在这个馏分收集器中,各种样品盘都可以用,样品瓶、多孔板、试管,以及新的漏斗盘。每个漏斗盘包含40个管路连接的漏斗(图2)。这些漏斗可以与任何容器相连,例如,大玻璃瓶。馏分收集器上最多可以安装三个漏斗盘,最多有120个回收区。 图2 安装在馏分收集器中的漏斗盘 结论 Agilent 1100系列纯化系统提供了三种回收收集的可能。最方便的办法是在馏分收集器废液管路中连接一个12位/13通阀。 第三方馏分收集器也可以通过BCD板外接点配置到系统中,由化学工作站进样程序控制。 最复杂的是在安捷伦化学工作站上将一个另外的Agilent 1100系列馏分收集器配置成回收收集器。另外,新的漏斗盘可以匹配样品瓶、多孔板或试管,对回收收集的体积没有限制。 ( Recovery Collection with the Agilent 1100 Series Purification System (用 A gilent 1100系列纯化系统进行回收收集)”, 发行号5988-9650EN。 ) 弥心平、尼莫地平和尼索地平都是带1,4-二羟基嘧啶结构的治心绞痛药物。在hypertonia, cardiacdisrythmia和angina pectoris情况下用做钙拮抗剂。从混合物中分离这三种药物是药物纯化的一个应用实例。本应用的目标是在一次运行中分别纯化出20mg各化合物,得到三个仅含目标产物的馏分。 结果与讨论 系统设置和配置 配置能操作普通方法的系统,21.2x50 mm Zorbax SB-C18柱,流速25 mL/min。在UV检测器后分流-大部分进入馏分收集器,分流部分进入MSD。由于样品溶解在DMSO中,所以设定的方法要让DMSO在任何感兴趣化合物之前洗脱出来。因此,在梯度开始之前,用水/乙腈90:10冲洗色谱柱2分钟。为了防止DMSO污染MSD, 在运行的前2分钟选择阀切换使液流进入废液瓶。 质谱引导的馏分收集 三个化合物的分子量分别为弥心平346.34、尼莫地平418.45和尼索地平388.42。所有三个化合物在正离子模式都显示有很强的碎裂。所以尼莫地平没有用其分子量418,而是用主要碎片质量342触发。图1显示了[M+H]+离子触发,质谱引导的馏分收集的结果。 馏分的再分析 为了检验纯化分离的效果,用一个事先用纯标准品建立的方法,在分析型液相色谱系统上对收集的馏分进行了再分析。各馏分的色谱图见图2。分析结果总结在表1中。弥心平、尼莫地平和尼索地平的纯化量分别为19.43 mg、19.05 mg和18.92 mg. 在25 mL/min流速下对三种药物进行了质谱引导的馏分收集。即使色谱柱高度过载,对收集馏分的再分析表明,在一次运行中可得到每种化合物20 mg, 具有高回收率,纯度达到了90%以上。这一结果说明配备了MSD的Agilent 1100系列纯化系统在高流速下具有卓越性能。 弥心平 尼莫地平 尼索地平 [mg] [mg] [mg] 馏分1 18.90 0.11 0.16 弥心平纯度 98.6% 馏分2 0.29 17.66 0.77 尼莫地平纯度 94.4% 馏分3 0.49 1.66 18.36 尼索地平纯度 89.5% 回收量[mg] 19.68 19.43 19.29 回收率[%] 101.3 102.0 101.9 表1 馏分再分析结果 图1 质谱引导的馏分收集结果 图2馏分的再分析 ( P urification of Pharmaceutical Drugs by Mass-based Fraction C o llection at Higher Flow Rates 55 (高流速下用质谱引导的馏分收集进行药物纯化),发行号5988-7113EN。 ) 随着对高纯度化合物的需求越来越大,合成化学家们通常都想更专注于有机合成问题,尽量少把时间花在对其合成产物的分析和纯化上。安捷伦主动分流器和延迟感应器作为安捷伦纯化/分析平台的组成部分,为用户提供了强大的纯化灵活性,最大限度地提高了效率,而且使用方便。 结果与讨论 提高纯化产率 主动分流器和延迟传感器在制药行业早期药物开发中非常有用。面对合成大量纯化合物的压力,有机和药物合成化学家们一直在寻找提高产率的工具。其主要目的在于: ·收集已知结构式和分子量的合成产物纯样品 ·收集微克到毫克级的组分 ·尽量缩短新化合物分析和纯化的时间 ·在收集样品方面,降低成本,缩短时间,减少工作量,简化技术 ·提高化学家和实验室的效率 图1为实现这些目标所用的系统示意图 灵活操作的主动分流器 这个系统中一个非常关键的组成就是安捷伦主动分流器。这个分流器的作用是从液相色谱液流中取出一小部分,转入质谱仪中。这一过程如图2所示。 主动分流器的主要性能如下: ·通过调节每个方法的分流比,可以改变液流的大小和切换速度。另外,主动分流器软件按照每个方法的设置,自动开启和关闭阀 ·不同的方法可以进行不同分流,不需要重新连接管路 ·不增加延迟,简化了色谱分析过程。反压最小,延长了系统组件的有效寿命 ·软件控制主动分流器参数和早期维护反馈(EMF),帮助制定更换密封垫的时间表 延迟传感器提高了系统准确度,而且设置方便 使该系统更简便可靠的另· 个重要特点是,能确定体积延迟。为了避免宝贵馏分可能的丢失,在质谱引导的收集系统中,感兴趣峰应在其达到馏分收集器之前先达到触发检 测器。这就需要正确安装管路,让每个检测器检测峰的时间与组分达到馏分收集器时间之间的延迟可以重复。为了更容易地实现这一点,馏分收集器中有一个内置的延迟传感器(FDS)。其dispensing臂的位置在传感器之上,可以精确计算出任何检测器与馏分收集器之间的延迟体积。 延迟传感器的主要优点如下: ·准确计算峰检测和馏分收集之间的时间延迟 。在方法和馏分收集器配置上自动校正延迟值 对于LC/MSD,延迟传感器将计算LC/MSD检测峰和峰达到馏分收集器之间的时间。和UV检测器不同,这种延迟受辅助泵流速和液相色谱泵流速的影响。因此,需要针对每个方法的流速进行重新校正。校正是用含染料和咖啡因的样品完成的,系统不安装色谱柱,将其一起进样。UV检测器和FDS对染料响应,而咖啡因m/z195离子用LC/MSD监测。监测延迟传感器的数据,并像任何其他2D检测器那样保存。 图1纯化流程图 结论 安捷伦主动分流器和延迟传感器可以大大提高从事化合物分析和纯化实验室的效率。主动分流器在优化分析和从不同基质中收集各种化合物时,具有更大的灵活性。延迟传感器简化了系统设置,确保了宝贵馏分的准确收集。 图2 主动分流器的操作 在理想情况下,从馏分收集器针尖洗下来的分析物组分正好相当于检测器信号所指示的组分,即,如果在馏分收集器针尖处记录--张色谱图的话,,它应该与柱后检测器测量的色谱图一致。但是,为了满足这一要求,应当考虑下列影响化合物回收率和收集可靠性的因素:延迟体积、扩散和系统响应。这里将概要介绍创新性的Agilent 1100系列纯化平台在设计上如何避免这些对色谱结果的影响因素。 结果与讨论 延迟体积校正 延迟时间是指分析物分子从检测器到馏分收集器所用的时间。为了精确触发馏分收集的起止,需要测定延迟时间。然后,可以将延迟时间方便地换算成与流速无关的延迟体积。通常,延迟时间的测定是用一种染料进样,记下其出现在馏分收集器针尖时的时间。这样测得延迟时间不仅费力而且也不精确。所以,Agilent 1100系列纯化系统包含创新性的延迟体积计算功能。这种专利能够全自动地精确测定延迟体积。 除了UV检测器以外,称为延迟传感器的第二个检测器也和馏分收集器组合在一起。无论什么时候当延迟校正剂注入流路中时,两个检测器都记录信号。两个信号之间的时间延迟(由内部计算减去和馏分延迟传感器之间的迁移时间差)即为延迟时间。根据校正过程的流速,系统自动计算出精确的延迟体积,并保存在馏分收集器内存中。现在系统可以计算出各流速下的校正时间,不需要重新进行校正。 扩散 化合物在从检测器到馏分收集器之间迁移时一个经常被忽略的现象就是扩散。扩散相当于使色谱峰变宽,从而严重影响色谱峰的分离度。根据Aris-Taylor方程,谱带扩宽与流速、管路长度和管径的4次方成正比。扩散效应对色谱峰的影响见图1。由此可见,检测器与馏分收集器之间连接管路的内径不合适,将影响纯化结果,如回收率低,甚至使化合物再混合。因此,宽流速范围的非特异性馏分收集器可能在高流速下能得到合格的结果(色谱柱内径大于25 mm),但在低流速下(色谱柱内径小于9 mm)将损 失回收率和纯度。而安捷伦提供了特别针对各种制备规模的馏分收集器。每一种类型的馏分收集器在生产时就为特定的流速范围和色谱柱内径提供了最佳性能,确保得到最大的化合物纯度和回收率。 系统集成智能化 系统集成智能化不仅让用户能从各种Agilent 1100 系列组件中配置适当的组件,而且还能进行实时的数据处理。实时数据处理在即时馏分收集和系统的安全可靠操作方面尤为重要,特别是在PC电源故障或网络中断的情况下。为了保证可靠而安全的操作,所有Agilent 1100灵活安全的系统操作都是通过控制器局域网(CAN)进行的。这样做的结果是系统操作完全不依赖PC。即使由于网络通讯阻滞、CPU被临时占用或完全丢失,导致系统和PC之间的通讯中断,馏分收集也将实时进行。因为这些馏分收集是精确地按照色谱图的提示进行的。除了CAN连接之外,各Agilent 1100系列组件还具有自己的智能系统,一旦系统接收到来自PC的任务,即被激活。PC仅仅是用户和仪器之间的接口。其功能简化为监测和评估实验结果。 结论 色谱纯化系统对目标化合物的纯度和回收率有至关重要的影响。通常要对纯化的样品进行再分析,以确认这些化合物的纯度。用户的经验显示,这些纯化样品的组成常常和相应色谱图中显示的有差异。洗脱液在检测器到馏分收集器之间迁移时很容易受到影响,这将严重影响收集馏分的组成。1100系列纯化系统有一些创新功能: 图1 管路内径对色谱分离度的影响,实验中样品体积、管路长度和流速保持恒定 ·分析物几乎不受干扰地转移到馏分收集装置 。专利的延迟体积校正,确保对检测器和馏分收集器之间的延迟体积进行精确测定,这对高纯度和回收率非常重要 ·精确的流路设计,确保低扩散,已经分开的峰不会重新混合,从而保证了高色谱分离度 ·安捷伦的系统集成智能化原理,为快速、可靠和精确的馏分收集提供了实时数据处理。 总而言之,安捷伦为宽应用范围提供了高性能的纯化系统,已经证明回收率接近100%。 安捷伦的1100系列馏分收集器设计的延迟体积最小,具有最好的馏分收集性能,这对低流速下的纯化尤为重要。图1是馏分收集器的示意图,有两个延迟体积V和VD2。 当检测出一个色谱峰时,馏分收集器必须等待这个峰从检测器的流通池到达分流阀,然后切换,收集这个馏分。因此,要把延迟时间t加到这个峰的起始时间t,上。为了确保所有峰都被收集,在峰结束时间t加上延迟时间t和tpe,馏分收集器切换回到废液位置。此时峰尾到达馏分收集器针尖。 结果与讨论 延迟体积Vp对回收率的影响 当一个峰在毛细管中移动时,由于流动相在毛细管横截面上的速率不同,将出现扩散。这是流动相和毛细管壁之间的相互作用造成。因此,扩散与毛细管的长度和内径有关。图2显示了随着延迟体积Vp的增大,色谱峰从检测器到馏分收集器的扩散。将馏分收集器的标准毛细管(0.25 mm 内径)换成长度不同但内径相同的毛细管,以增加V。 根据检测器信号触发馏分收集,即馏分的峰宽由色谱峰的宽度决定。当色谱峰到达馏分收集器时,由于扩散峰将变宽,但切换到收集位置只是根据检测器峰宽测定的时间窗口。这将导致在色谱峰的开始和结束时化合物丢失。延迟体积V,,越大,化合物的丢失越多。Agilent 1100系列馏分收集器最大限度地降低了这种影响,因为它设计了最小的延迟体积,建立1100系统后还可以再切割馏分收集器的入口毛细管,尽可能使其变短。 延迟体积Vp对分离度的影响 对已分开峰再混合的影响因素中,扩散也是一个重要的方面。测量两个峰分离的参数是用5o计算的分离度。之所以选择这个方法,是因为它的计算是根据5o峰高(峰高的4.4%)处的峰宽,接近峰的基线。 为了表示增大延迟体积V对分离度的影响,用检测器,也在馏分收集器处检测两个色谱峰。通过增加两个不同内径(0.25mm和0.8mm)的管路增大延迟体积。然后比较这两个峰在馏分收集器和检测器处的分离度,得到相对分离度。图3显示随着延迟体积V,的增大,相对分离度降低。也说明延迟体积相同时,随着毛细管内径的增大,相对分离度减小。这是由于毛细管横截面对分离度的影响,在横截面的计算中半径进行了平方。 图1 结论 在本技术报告中,我们报道了不同延迟体积、检测器信号延迟的影响,以及如何自动测定延迟体积。 用不同延迟体积触发峰得到最大回收率。最大限度地缩小延迟体积以避免低回收,或扩散导致的已分离峰再混合。 图2 通过增大毛细管体积增加扩散 图3 两种不同内径毛细管的相对分离度 ( 66 P eak-based Fraction Collection with the Agilent 1100 Series Purificati o n System AS -Influence of Delay Volumes on Recover y ” (Agilent 1100纯化系统AS基于色谱峰的馏分收集-延迟体积对回收率的影响),发行号5988-5746EN。 ) 应用 技术 纯化策略 阀的应用 基于第三方检测器的色谱峰进行馏分收集 蒸发光散射检测器(ELSD)是制药行业常用的一种检测器。它可以检测没有发色团的混合物,检测器的响应只与洗脱化合物的量有关。因此,非常适用于纯度检查。 系统设置可以将蒸发光散射检测器以及其它非安捷伦检测器安装在1100系列纯化系统中。将4种糖皮质激素标准品的分离作为实例。对收集的馏分再分析以说明系统的纯化性能。 设备 系统的设置和配置 因为 ELSD是有损检测器,通过一个分流器(安捷伦1:10/1:20分流器)设置在流路中。主要进入馏分收集器,分流进入ELSD(图1)。 为了让ELSD信号触发馏分收集,将检测器的模拟输出连接到一个通用接口盒(UIB)上。UIB再通过CAN连接到1100系列组件上(图2)。任何其它带模拟输出的非安捷伦检测器都可以按这种方法设置在安捷伦1100系列纯化系统上进行基于色谱峰的馏分收集。 蒸发光散射检测器的延迟体积校正 要准确收集馏分,必须测定ELSD和馏分收集器分流阀之间的延迟时间。用馏分收集器内置的安捷伦馏分延迟传感器可以做到这一点。Agilent1100系列纯化系统用户指南中记述了延迟校正的步骤。可以使用标准的延迟校正试剂(G1946-85020)和化学工作站的标准延迟校正方法。因为分液流中的化合物绝对应该在主液流到达馏分收集器之前就到达ELSD, 所以,延迟体积必须是正值。如果测得负的延迟体积,化合物在达到ELSD之前先进入了馏分收集器,那么在触发前都将流入废液瓶。为了避免出现负的延迟体积,分流器和ELSD之间的管路连接必须尽可能短,并使用小内径毛细管。如果不可能缩短进进ELSD的毛细管,只能延长从分流器到馏分收集器的毛细管。但是,这样做将增大扩散,从而导致馏分收集性能降低, 也应该避免。 结果与讨论 蒸发光散射检测器信号触发馏分收集要用蒸发光散射检测器信号触发馏分收集,在化学工作站上的 SetupFraction(Collector窗口中必须将UIB选择在PeakDetector。像任何UV检测器一样可以设置UpSlope(正斜率)、Doun Slope(负斜率)和Threshold(阈值)等参数。分离糖皮质激素时,从表中去掉了正负斜率值,只用70 mV阈值触发。图3显示了糖皮质激素标准品的DAD、双通道A/D接口和UIB信号-竖线标志着收集的馏分。 为了确认纯化结果,不用分流器在同一系统上对馏分进行再分析。结果清楚地表明,四种化合物可以分开,不含任何杂质。说明Agilent1100系列纯化系统使用非安捷伦蒸发光散射检测器具有卓越的性能。 结论 在这个例子中,使用Sedere SedexModel 75 ELS检测器触发基于色谱峰的馏分收集。但也可以用同样方法设置任何其它非安捷伦检测器,只要是模拟输出。作为一个例子,我们用分析型制备液相色谱分离了四种糖皮质激素标准物。 图1 在流路中用分流器设置ELSD 图2 使用UIB和双通道A/D接口盒的纯化系统的线路连接 图3 基于ELSD信号进行留分收集 ( P eak-b a sed Fraction Collection Using an Evaporative Light Scattering Detector with the Agilent 1100 Series Purification System”(用安装蒸发光散射检测器的Agilent 1100系列纯化系统进行基于色谱峰的馏分收集),发行号5988-5816EN。 ) 在药物研发中,要进行活性检测需要对化合物进行纯化。由于合成和筛选通量的增加,传统的低通量纯化技术,如制备TLC或结晶,成为合成实验室的瓶颈。全自动纯化的现代技术是制备液相色谱。在药物研究的早期阶段,通常要少量合成许多种化合物。因此,纯化可以在5 mm或更小内径的色谱柱上进行,称为分析型制备液相色谱。但在药物研发的后期,化合物的纯化量和色谱柱内径将增大。 安捷伦提供1100系列纯化系统PS制备泵,最大流速100 mL/min, 最高反压400 bar虽然该系统和泵都是为高流速而开发的,但在5 mL/min以下的流速也可以使用,因为安捷伦1100系列制备泵具有卓越的性能。 在本应用报告中,我们报道了不改变安捷伦1100系列纯化系统PS的配置,用0.35 mL/min流速在3 mm内径柱和100 mL/min在50mm内径柱上对一个模型样品的梯度分析。 结果与讨论 分析型方法开发 在Agilent 1100系列系统上建立了三个模型化合物的分析方法,该系统包括构成所需梯度的两个Agilent1100系列制备泵。采用3.0-mm内径的ZORBAX SB-C18柱,流速0.35 mL/min。图1中的色谱峰显示了良好的分离度和峰形,证明Agilent 1100系列制备泵即使在低流速下也具有良好的性能。 放大计算 根据图2中的公式计算出整个放大过程。用上面的公式计算流速,将3.0 mm内径柱流速0.35 mL/min选做起点。用下面的公式计算进样体积。考虑到流速较高和加载的样品量,对于更大的色谱柱需要将3-mm光程长度的不锈钢制备流通池更换为0.3-mm光程的石英流通池。这将使进样体积的计算值增大10倍。计算出的所有色谱柱和流通池的流速和进样体积总结在表1中。 x=柱1进样体积 x。=柱2进样体积 r=柱1半径 r,=柱2半径 C=柱长比=1 图2 放大计算所用的公式 分析和制备型纯化 在四种不同色谱柱上(4.6mm内径、9.4mm 内径、21.2 mm 内径和50 mm 内径)进行分析和制备型分离,采用的流速和进样体积列于表1。表3中的结果色谱图说明了两点-第一, Agilent 1100系列制备泵在很宽的流速范围内都具有良好性能。第二,在ZORBAX柱上可以放大,不会损失分离度,这一点对快速放大方法(不需要对方法进行再开发)非常重要。 结论 Agilent 1100系列制备泵在0.35-100 mL/min流速范围内显示出了卓越的性能。 ·该泵与化学工作站的OQ/PV和EMF(早期维护反馈)等特性完全兼容 。可以将在3mm内径的ZORBAX柱上的应用放大到50 mm内径的柱上,对分离度没有任何影响。通过缩短重新开发或调整方法所需的时间提高了分析通量 图1 0.35 mL/min流速下的色谱图 图3 放大计算的结果 柱 尺寸 流速 进样体积 检测器流通池 1(图1) 150x3.0 mm 0.35 mL/min 0.8 pl 3 mm SST 2(图3) 150x4.6 mm 0.85 mL/min 2.0 pl 3 mm SST 3(图3) 150x9.4 mm 3.5 mL/min 80 pl (8 plx10) 0.3mm石英 4(图3) 150x21.2mm 18 mL/min 400 ul (40 plx10) 0.3mm石英 5(图3) 150x50 mm 100 mL/min 2200 ul (220 ulx 10) 0.3mm石英 表1 不同柱尺寸的流速、进样体积和检测器流通池 流速从1到100 mL/min的制备泵性能 Agilent 1100系列制备泵是一种等度高性能泵,有两个平行活塞。不需要更换泵头,最高流速可以达到100 mL/min, 压力400 bar。两个泵通过低死体积混合装置连接,形成高压梯度混合系统,内部延迟体积只有0.7mL左右。泵的其它性能包括用蠕动泵自动清洗密封垫和自动电磁吹扫阀。虽然该泵系统是为最高流速100mL/min而设计的,但在较低流速下也显示了非常好的性能。 注: ( 所有实验都在 一 套用于安捷伦实验室日常工作的系统上进行。由于您 系统运行的时间不同,测试结果可 能略有差异。 ) 结果 1.流速精密度-等度 流速精密度对每次运行之间保留时间的精密度非常重要。由于泵的流速通过制备泵固件的补偿计算控空,流速精密度由反压决定。 流速精密度测试设置 ( ·流速精密度通过收集5分钟的溶剂量测定。通过称量收集的水,并除以密度,测出收集的体积 。测试在25 mL/min流速下进行 ) ( ·在 三 种不同反压下测定精密度 (< 1 00 bar , > 100和<200 bar , > 200 bar) ) ( ·在每个压力下运行5次测定精密度 ) ·所有实验都用限流毛细管代替色谱柱 流速精密度测试结果 图1显示了等度流速精密度测试结果。标记了流速精密度的相对标准偏差。不同反压的相对标准偏差都低于0.3%。 流速精密度测试设置 ·流速精密度通过收集7分钟的溶剂(水、甲醇)测定。测定收集的体积 ·测试在10 mL/min流速下进行 。梯度:100%水1分钟,5分钟之内从100%水到100%甲醇,100%甲醇1分钟 ·在三个不同的反压范围内测量精密度 ·在每个压力下进行5次测定精密度。所有实验都用限流毛细管代替色谱柱 流速精密度测试结果 图2显示了梯度流速精密度测试结果。用黑色误差棒显示流速精密度相对标准偏差,不同反压设置下的偏差均低于0.4%。 结论 ·证明了在0.35到100 ml/min流速范围内1100系列制备泵的性能 ·在5 ml/min流速以下性能几乎与分析泵(如1100系列四元泵)样好 ·毛细管,特别是较高流速所需要的大内径毛细管,将降低系统的整体性能 因此安捷伦科技公司提供的专业系统,从毛细管到制备级,而不是用一个系统包含全部流速范围。对特珠应用确保最佳性能,而不是在宽应用范围内都是低性能。 图1不同反压下等度流速精密度的相对标准偏差 图2 不同反压下梯度流速精密度的相对标准偏差 ( "Performance C haracteristics of t h e Agil e nt 1100 Series Preparative Pump” (Agilent 1100系列制备泵的性能特征), 发行号5988-7110EN。 ) 在现代药物研发实验室中,需要分析和纯化的新药化合物数量急剧增加。合成有机化合物和药物的化学家们经常要“走到”LC/MS系统中心,提交样品,进行分析和纯化,然后返回其实验室再继续合成。化学家们越来越需要可以在自己实验室内自己的PC机上浏览数据,而不需要返回到LC/MS中心实验室去。 新的安捷伦化学工作站数据浏览器使远程数据浏览更容易而有效。在系统上获取原始数据,产生一个中间文件(称为.AEV文件),然后把 这个.AEV文件能够通过服务器或电子邮件传送到远程PC上。虽然目标是药物研发,但任何需要浏览LC或LC/MS化学工作站系统数据的实验室都需要浏览器。 数据浏览器的主要功能 浏览器在药物研发实验室中的主要用途是尽可能快地回答大多数样品的下列问题: 我得到了想要的化合物吗?如果答案是肯定的,那么纯度大约有多少? 主屏幕包含了一系列了视窗。所显示的内容和应用密切相关,也可以由用户定制。图1显示了药物研发实验室浏览含目标化合物反应混合物微孔板的典型配置。在屏幕左下角的板浏览视窗中,用户可以观察微孔板上或其它1100系列自动进样器盘上(如100位盘)样品瓶位置,图形表示了样品。 图1 化学工作站数据浏览器主屏幕 在图1中,绿色孔表示找到了目标质量,红色孔表示按照设定的阈值没有找到目标质量。用户只需点击相应的孔,就可以看到更多的样品信息,如UV和MS色谱图。如果对样品进行了纯化,馏分信息也将显示在馏分表中。 下列关键问题是数据浏览器可以回答的,将有助于下一步药物开发做出正确决策: 另外,数据浏览器还可用于: ·提供峰纯度信息(共洗脱组分的数目) ·同时从板视窗中浏览所有数据 ·同时在屏幕上各独立的窗口中比较多个样品 图2 。提供用户定制的灵活报告 通过数据浏览器软件定制报告的实例 报告 数据报告是针对药物研发的另一个重要功能。有了灵活的化学工作站数据浏览器,用户可以通过选择报告中的项目来优化这项功能。图2是一个报告的实例,显示了药物开发中确认目标化合物是否存在的关键项目。 ( Enhancing Lab Productivity with the Agilent ChemStation Dat a Browser (用安捷伦化学工作站数据浏览器提高实验室效率),发行号5988-8490EN。 ) 应用 技术 纯化策略 阀的应用 纯化常用的梯度是有机相从5到95%,但这有时并不能给目标化合物带来足够的分离度。为了提高分离度,还要对方法进行优化,在目标化合物出峰的洗脱液比例附近使用更平缓的梯度。根据保留时间窗口(预制备分析中化合物的洗脱时间)选择制备方法。 结果与讨论 为了设定方法的保留时间窗口,先用分析方法测定几个标准化合物的保留时间。生成一个图表说明这些化合物的保留时间和洗脱液实际组成之间的关系。任何一个化合物,只要已知它在分析分离中的保留时间,就可以从这个图表中提取或计算出洗脱液的组成。用同样的方法也可以确定保留时间窗口的起止梯 度组成。本应用报告提供了8种安捷伦标准品的洗脱组成,用简单的四个步骤生成一个制备方法: 1.用分析方法等度测试样品和电喷雾LC模型样品 2.用测定的保留时间和已知的梯度洗脱组成生成图表 3.从图表中计算或提取出起止梯度组成,以优化制备方法 4.为各化合物制备方法设置专用的梯度 纯化实例 为了纯化目标化合物磺胺2,用普通梯度和优化的梯度在制备柱上对样品进行了分离。结果见图1(A,B)。 用普通梯度目标化合物的一部分与前一个峰一起洗脱,但用优化的方法达到了基线分离。磺胺2与前面峰的分离度几乎提高了2倍,磺胺2与后面峰的分离度提高了大约50%。 结论 图1 在本应用报告中,介绍了根据预制备分离的时间窗口得到优化的制备方法。使用等度测试样品和电喷雾LC标准样品,在不到1小时的时间内,用简单的4个步骤完成了整个过程。应用实例说明用优化梯度可提高目标化合物与相邻组分之间的分离度。 用普通(A)和优化梯度(B)进行制备分离 在本应用报告中,我们报道了针对针对药物化学类别制定化合物纯化策略药物化学类别制定纯化策略。制定 这种纯化策略的目标如下: ·简单的自动工作流程 。为化学家们提供一套样品制备和方法选择的指导方针 问题样品的纯化-不溶性样品、结果与讨论极性样品和弱电离的样品 ●可以随意输入馏分的起始位置(如果没有输入位置,将自动使用馏分收集器下一个位置) 合作者Darryl McConnell, GeorgEgger, Boehringer Ingelheim,Austria 不溶性化合物的进样 为了在梯度开始条件下将不溶性化合物进样到流动相中,,可以使用两种进样技术:有机相进样或夹层进样。 极性化合物的纯化 为极性化合物设定了专用方法。为了避免极性化合物与DMSO峰一起洗脱,要采取下列措施: ·将梯度起始条件设定为5%乙腈(标准方法用的是10%) ·梯度起始条件维持2.5分钟。将梯度终止条件设定为50%乙腈 (标准方法用的是95%) 本文报道了一个实际反应混合物的纯化。在Agilent 1100系列分析系统上对粗反应混合物进行了分析,结果显示所存在的目标产物是一对顺反异构体混合物。分析色谱图见图1。 为了分离这两个异构体,对粗产物进行了再纯化,这次使用的是UV信号和质谱信号的逻辑“与”进行收集。如图2所示,采用这个方法,成功地将异构体收集到了两个馏分中。 结论 本应用报告报道了针对药物化学类别制定化合物的纯化策略。建立了一个制备型液相色谱-质谱简便的自动化纯化流程。针对不同样品类型生成了一套5种预先设置的方法,标准方法适用于日常样品的80-90%。进一步建立了针对不溶性样品、极性样品和弱电离样品的方法。UV信号和质谱信号用逻辑“与”的馏分收集建立了另一种方法。 图1 粗反应混合物的分析色谱图 图2 用UV信号和MS信号的逻辑“与”进行馏分收集 应用 技术 纯化策略 阀的应用 用下列三种馏分收集策略可以对进样样品中进行多种或单一馏分的完全或低回收收集。但是,在所有这三种情况下,都不需要对馏分再分析以鉴别想要的目标化合物。化合物鉴定可以通过将1100系列质谱检测器连接到Agilent 1100系列纯化系统上实现。 结果与讨论 在本应用报告中我们介绍了下列三种纯化策略: 1.用MSD监测基于时间的馏分收集 2.用MSD监测基于峰的馏分收集 3.质谱引导的馏分收集 1.用MSD监测基于时间的馏分收集 用基于时间的馏分收集得到馏分,保证进样的样品几乎没有丢失。 基于时间收集馏分,在2到7.5分钟之间收集到了80个馏分,结果见图1中的色谱图。指示了收集馏分的起始标志。 在配备质谱检测器的系统上进行化合物纯化 为了鉴定含有目标化合物(目标质量数为241 amu)的馏分,将质量数242([M+H]+)的提取离子流图(EIC)与总离子流图(TIC)进行重叠比较。图2显示,在多孔板B-02到B-06位中含有目标质量数。 2.用MSD监测基于峰的馏分收集 用第二个策略,基于阈值设置为50 mAU的UV信号进行馏分收集。确保样品中的主峰被收集,与基于时间的馏分收集相比,所收集馏分的数量明显减少。 为鉴别含目标化合物的馏分,还把质量数242([M+H]*)的EIC与 TIC图进行重叠比较。 当使用质谱引导的收集馏分时,只有含目标质量数的峰进入MSD, 这个目标质量的EIC响应强度超出设定阈值时,才触发馏分收集。确保在大多数情况下,每次样品运行中只有含目标化合物的组分被收集。缺点是大部分进样的样品都没有回收。质谱引导的馏分收集结果见图3。 如图3所示,只有在1号瓶中的馏分含有目标质量数,所以收集的质量数减少到1,但样品损失增加到了95%。 结论 虽然基于时间或色谱峰的馏分收集不需要MSD, 但有了MSD的优点是不需要通过繁琐的再分析测定含有目标化合物的馏分。需要的馏分可以很容易地通过将TIC与目标质量的EIC重叠进行鉴定。 图2 TIC和EIC重叠鉴别目标馏分 图3 质谱引导的馏分收集结果 下面将介绍四种分离策略: 对未达到基线分离的化合物进行纯化 1.通过高阈值设定进行基于峰的馏分收集 2.用正负斜率进行基于峰的馏分收集 3.通过峰的时间切割进行基于峰的馏分收集 4.质谱引导的馏分收集 结果与讨论 1.通过高阈值设定进行基于峰的馏分收集进行基于峰的馏分收集最简单的方法,是只根据阈值收集。即,只要检测器信号超过预先设定的数值馏分收集就开始,信号低于设定阈值时收集就终止。这种基于峰的馏分收集适用于大多数纯化应用。未基线分离峰也可以分开,但阈值必须设置为相当高的数值。这样做将导致峰开始和结束部分的化合物不被收集,从而降低回收率。 2.用正负斜率进行基于峰的馏分收集 为了避免峰开始和结束部分化合物的丢失,安捷伦化学工作站和纯化/高通量软件为基于峰的馏分收集提供了两个应用参数-正斜率和负斜率。设定这些参数后,如果正斜率和阈值两个都超出,触发峰收集的起点。如果信号满足一个指标,即,如果低于阈值或低于负斜率值,则触发峰的终点。结果是可以将阈值设置在相当低的数值,使两个未基线分离的峰分到两个馏分中,而不丢失峰的起点和终点。 3.通过峰的时间切割进行基于峰的馏分收集 从上可见,使用正斜率和负斜率进行基于峰的馏分收集,获得了良好的回收率和纯度。但是如果重叠的峰很宽,因而重叠面积很大,将使收集的馏分明显不纯。在这种情况下,更好的触发方法是基于信号开始收集馏分,然后基于时间收集,将两个峰切割成若干部分。要实现这一点, 可以使用安捷伦化学工作站软件Setup fraction collector(设置馏分收集器)窗口下Auxiliary部分的Max. fill volume perlocation(每个位置的最大充人体积)功能。使用了这个功能,方法中预先设定的填充体积将被改变。 在图2显示的例子中,将深孔板作为馏分收集容器,设定填充体积为2 mL。因为所用的流速是20 mL/min,每6秒钟充满一个孔。两个峰的总峰宽大约45秒,需要8个孔。为了避免馏分收集针头移到下一个位置时丢失化合物,将馏分 CollectionMode(收集模式)设置为Continuosflow。在这种模式下,如果用多孔板作为馏分收集容器只能这样配置,收集针头移到下一个位置时,分流阀不切换到废液位置。馏分收集结果见图2。 4.质谱引导的馏分收集 另一个从未基线分离峰中纯化化合物的方法是质谱引导的馏分收集。因为触发是根据最大丰度目标质量数进行的,一旦在MS图中第二个峰的目标质量变成主要离子,即启动第二个馏分的收集。馏分收集的结果见图3。收集馏分再分析显示,基于MS的馏分收集回收率和纯度结果与用正负斜率基于峰的馏分收集结果相似。 结论 只根据阈值基于峰的馏分收集是分离两个化合物的最简单方法,但必须设较高的阈值,这可能导致峰起始和终止部分的化合物丢失。用化学工作站软件的正斜率和负斜率功能可以避免这一问题。如果这两个峰很宽,并在较宽的区域有重叠,可以使用基于峰的馏分收集,并将峰进行时间切割。这需要对正确的馏分收集体积进行一些手工计算,馏分数目将较多。如果纯化系统配备了MSD,可以进行质谱引导的馏分收集,也只能得到两个馏分。 图2 采用时间分割的色谱峰引导的馏分收集 图3 应用 技术 纯化策略 阀的应用 什么是正负斜率?如何用它决定馏 基于色谱峰的馏分收集中正负斜率的应用分触发?何时需要使用正负斜率? 1.用“正负斜率”决定馏分触发 2.应用 尖锐峰和扁平峰的分离 通过使用正负斜率,可以从扁平峰中分离出尖锐峰。图3表明,很容易从扁平的峰中分离出较尖锐的峰。要做到这一点,必须在馏分收集开 始之前就已知正负斜率值。这只能通过试运行完成。 未达到基线分离的化合物的分离 如果两个峰没有基线分离,有几个策略将这两种化合物收集到不同的馏分中,并具有良好的回收率和纯 用正负斜率决定馏分触发 图2中的色谱图对馏分收集的起止做了标记。当超过阈值和正斜率设定值时(标记1),触发馏分起点。当信号降到阈值以下或符合负斜率标准时,触发峰终点。在标记2,馏分终止,因为在局部最小处斜率等于零,满足了负斜率指标。在这一个点上阈值仍然大于设定值。在标记3,正斜率再次超出,阈值仍高于设定值。最后在标记4,信号已降到阈值以下,因此,不管负斜率值是否符合标准,馏分收集都终止。 图1 斜率是一个峰的-阶导数 度。其中之一是用正负斜率进行基于峰的馏分收集。因为在两峰之间的斜率最小,等于零,因此即使信号没有低于阈值,也可以被分到两个馏分中。 基线漂移色谱图中化合物的纯化 对基线漂移色谱图进行基于峰的馏分收集,不能只根据阈值进行馏分收集。如果基线向上漂移,高于阈值上的所有部分都将作为馏分被收集。如果基线向下漂移,只有峰的一小部分,或者根本没有被收集。因此必须采用正确的正负斜率值。图4显示了对基线上漂的色谱图进行基于峰的馏分收集。 结论 正负斜率可用于触发基于峰的馏分收集。明显的优势在于能够设定两个参数,,而不是用一个斜率参数。对于许多应用来说,只用阈值就足够基于峰进行馏分收集,但我们也列举了一些需要正负斜率的应用实例。 ( Sophisticated Peak-based Fraction Collection- Working with Up and Down Slope (基于色谱峰的馏分收集的高级功能-正负斜率的应用),发行号 5 989-0511EN。 ) 图3 a)正负斜率:30mAU/s, b)正负斜率: 120 mAU/s c)正负斜率:250mAU/s, d)正负斜率: 400 mAU/s 图4 阈值设置:15mAU, 正负斜率: 15mAU/s 应用 技术 为什么在样品工作流程的后续阶段用专用液相色谱上进行纯度分析比馏分收集后再分析检查纯度比直接进行更好?这是根据1100系列纯化系统用户的经验得出的结论。 结果与讨论 一般工作流程 在药物研究,以及其它行业(如Crop Science)中,寻找活性化合物的一般工作流程可以概括为6个步骤: 1.由化学家提交不纯的样品 2.在制备液相色谱系统上进行纯化 3.馏分收集 4.溶剂蒸发,化合物称量 5.纯化合物再溶解配制成一定浓度 6.拿出少量溶液进行活性试验 在这一工作流程中的某一步,应检测提交进行活性试验的化合物(步骤6)具有需要的纯度,通常必须大于90-95%。 图1 1.浓度梯度 在一定时间内从色谱柱洗脱出的目标化合物在馏分中形成一个浓度梯度。在馏分容器中可以看见这个浓度梯度(图1)。如果直接从试管中吸出小体积进行再分析,那么这个样品肯定不能代表进行活性试验的样品。 2.结晶 为了更好地溶解,提交进行纯化的样品经常不是用流动相作为溶剂,而是用更强的溶剂如DMSO或DMF.然而,反相色谱中所用的流动相通常是水和乙腈或甲醇。取决于样品的进样量和溶解度情况,目标化合物离开流动相开始结晶。如果进行纯度检查的样品是取自母液,那么这个样品同样也不能代表活性试验的样品。 3.蒸发过程中的分解纯化后,需要对含目标化合物馏分的流动相进行蒸发。可以通过真空离心或加热和氮气吹干蒸发溶剂。称量残渣后,用适当溶剂(例如DSMO或DMF)再复溶,配制成一定浓度。如果直接从馏分容器中取样检测目标化合物的纯度,就无法检测目标化合物在蒸发过程中可能的分解。最坏情况下可能错过高活性化合物。 哪一步是纯度再分析的合适步骤? 在工作流程中进行再分析,而不是馏分收集后直接分析,在前面章节中提到的三个问题都不会发生。因为复溶化合物的一部分用作活性试验(步骤6),同一溶液的第二部分可用于分析和纯度测试。这绝对能够保证是对活性试验的代表性样品进行再分析。 自动化馏分再分析是通往完全自动化纯度解决方案的另一个步骤。结果表明: ( 自动化的增加牺牲了测量结果的可靠性 ) ( ·由于浓度梯度和在馏分容器中可 能的结晶,可能导致错误的纯度测定结果,还可能因为蒸发过程 中的分解,错过了活性化合物 ) ( 如果在样品纯化和活性试验的最后一步,用专用的分析型液相色谱系统进行分析,所有这些缺点都可以 克服。 ) 下面介绍了两种增加色谱柱载样量的方法-夹层进样和有机相进样。 高浓度样品的进样 结果与讨论 夹层进样 夹层进样是将样品夹在两个样品溶剂层之间进样。例如,如果该化合物是溶解在DMSO中,那么就在样品的前后放入两段DMSO(图1)。当整个夹层开始和流动相混合时,混合的起始部位在前段溶剂,那里样品浓度为零,所以不可能发生沉淀。在安捷伦化学工作站上通过使用进样器程序,可以很容易地实现 图1夹层进样 夹层进样。 夹层进样的优点 ·样品在达到色谱柱之前不会与流动相接触,,因此,在管路的重要部位不会发生沉淀 ·不需要再配置其它硬件。夹层进样可以在标准系统上用进样器程序完成 夹层进样的缺点 ·样品必须以相当窄的条带保留在 柱上。强的样品和栓塞溶剂将导致 在柱头出现明显的样品谱带变宽·如果溶剂栓和样品体积相对于柱体积的比例过大,部分化合物将随着溶剂前沿被洗出 ·栓塞和样品溶剂可能破坏固定相的平衡 有机相进样 有机相进样需要对制备泵和进样器管路进行改造。其思路是把进样器连接在有机相输液泵之后,在水相和有机流动相混合之前(图2)。即,样品在到达混合三通和预柱前,只接触有机相。因此,在自动进样器切换阀等重要的管路部位,不会发生沉淀。三通必须靠近预柱,从而使沉淀发生之前样品已经进入到柱头。 有机相进样的优点 ·不需要很强的样品溶剂,那些溶剂可能会导致峰变宽和色谱柱 ·因为Agilent 1100系列梯度版的制备泵包括两个物理泵,梯度程序非常容易。可以和标准配置的设置方法一样,不需要流速梯度程序 有机相进样的缺点 ·虽然在自动进样器阀这一管路中的重要部位不会发生样品沉淀,但混合三通或预柱柱头仍有可能因沉淀被堵塞。虽然柱头沉淀一般只会导致压力升高,但混合三通的堵塞是很严重的问题 ·设置有机相进样的纯化系统,需要对系统管路进行改造。即,在硬件没有改动的同一个系统上,不可能同时进行有机相进样和标准进样 ·使用夹层进样,将样品放在两段纯溶剂之间,避免样品与梯度起始流动相发生混合。用安捷伦化学工作站进样器程序可以很容易完成 ·进行有机相进样时,自动进样器要连接到在与含水流动相混合之前的有机流动相管路中。即,混合和沉淀不会发生在自动进样器阀上,而是发生在不会出现问题的与自动进样器非常靠近的预柱柱头 随着过去几年来合成有机化学的飞速发展,更需要有简便、耐用、快速的系统,对数量巨大的新化合物进行分析和分子量确认。合成化学家很想把精力集中在有机合成问题上,尽量缩短用在化合物分析上的时间。这里介绍的Agilent LC/MSEasy Access软件,让用户只需要轻松地处理样品,输入简要的样品信息,从列表中选择方法、由系统指定样品的位置,然后返回自己的实验室,等待结果的电子邮件。 LC/MS Easy Access软件的主要特点 ·现在用化学工作站数据浏览器可以浏览电子邮件数据 ·无需重新设置馏分收集器,因为在系统运行时馏分可以不断取走 ·样品提交和状态检查非常简便。快速确认分子量和目标离子的存在 ·支持Agilent 1100系列多孔板进样器和自动液体进样器 ·方法改变时自动预平衡 ·支持自动化的电子邮件传递数据和报告 ·灵活的管理工具,设置用户进入、列队等候和项目管理 ·多台仪器网络化从而使仪器可以分享数据库,不需要为每台仪器管理多余的配置,减少了管理任务 图1 系统提示用户在哪里放置样品,提供大约完成时间 图2 结果与讨论 用户输入密码登录后(可选择安全性),描述样品、从列表中指定方法,系统显示进样器的哪些位置应该放置样品(图1)。图2显示了系。当前的样品和序列中还要等候的 统的整个状态,包含如下重要信息:大约时间。当前运行的方法和最后提交者的 姓名 ·化学工作站、自动进样器和UV/Vis灯的状况 系统管理 系统管理员对整个Easy Access系统的管理负责。主要权限包括: ·用户和组别管理,包括选择密码、方法进入和化学工作站的使用 ·样品序列管理,包括将优先样品移到队列的前面 。方法管理,指定用户可用的方法 由电子邮件传送结果 系统可以指定将分析结果通过电子邮件发送给选定的用户。右侧的例子是一个接收到的信息。另外,电子邮件中还可以包含附件报告,显示找到的目标质量、色谱图和质谱图。 Your sample run has beencompleted. (您的样品已经做完) Sample:ESdemo05' (样品:。......) Info: Sulfa Drugs' (信息:磺胺药物) 图3确认目标质量m/z270是否存在的多页报告中的一页 LC/MS Easy Access是一个非常有 效的工具,帮助合成有机化学家提 高效率,在更短的时间内,需要较 少的分析专业知识,就能确切鉴定 其合成化合物。 Increasing Productivity with LC/MS Easy Access Software"(用LC/MS Easy Access软件提高效率),发行号5988-5525EN。 应用 技术 纯化策略 阀的应用 在制药行业化合物纯化中,立体异 构体、非对映异构体,特别是对映 异构体的分离非常重要,但是很有 挑战性。经常只能对这些化合物达到部分分离。使用更长的色谱柱以得到更好的分离度,常常又受到这结果与讨论些色谱柱产生较高反压的限制。因此,开发出了几个循环技术,将化 工作原理 合物送入同一根色谱柱若干次,直到获得足够的分离度。在本应用报告中,我们介绍了用Agilent 1100系列纯化系统如何进行循环色谱。 被保留在柱2上。当这些化合物在柱2被进一步分离后,用切换阀再将柱1接到柱2后面。化合物从柱2洗循环色谱技术的工作原理是将色谱 脱下来后,再被保留在柱1上,柱2柱接到流动相的液流方向上(图1)。 又接到柱1后面,如此往复进行。化合物进样后,从柱1中洗脱出来, 图1 色谱结果 增加有效色谱柱数量的色谱结果见图2。如图所示,这些化合物用一根色谱柱很难分开,但通过6个虚拟色谱柱,4次阀切换,达到了基线分离。 局限 ·循环色谱只适用于分离保留时间相似的化合物,如,等度洗脱分离立体异构体、非对映异构体和对映异构体,不适合梯度分离 ·循环次数受两个化合物在一根色谱柱上的驻留时间限制 纯化实验 图2 用循环色谱技术,相当于用6个色谱柱,馏分收集得到两个纯度100%的馏分,见图3。 各种数量虚拟色谱柱的分离度 结论 在本应用报告中,我们报道了用2位/6通阀在Agilent 1100系列纯化系统上进行循环色谱,分离立体异构体、非对映异构体和对映异构体。 图3 用循环色谱6个虚拟色谱柱进行基于色谱峰的馏分收集 ( Recycle Chromatography with the Agilent 1100 Series P u rification System (Agilent 1100系列纯化系统上进行的循环色谱), 发行号5989-1693EN。 ) 制备液相色谱中的交替柱再生可以用2位/10通阀完成。 制备液相色谱中的交替柱再生 阀的配置、毛细管连接和泵的通用方法设置,内外阀都相同。后面不同的只是软件,将在下面介绍。 结果与讨论 阀的设置 外部2-位/10-通阀从图1显示的SetupValve窗口控制。要进行自动化柱再生,必须检查Next position afterrun框。自动将Position设置在Usecurrent。用这一设置在运行完成后自动将阀切换到下一个色谱柱,运行开始时保持在这个位置。要了解更多信息,例如,毛细管连接的配置等,可以选择Help查找。 序列运行 没有柱再生,纯化运行的步骤“吸取与进样”、“梯度运行”、、“柱清洗”和“柱平衡”等将按顺序运行,见图2。在这个应用报告中,方法总的周期时间是13分钟。 用等度泵进行交替柱再生 用梯度泵进行交替柱再生 如果用等度泵进行交替柱再生,下一步纯化在第二根色谱柱上开始后,可以进行“柱平衡”步步(图3)。为了确保泵和阀之间的体积都充满梯度起始比例的流动相,柱清洗后还要一段有额外的冲洗时间。这段时间的长短取决于系统设置,例如,自动进样器的延迟体积。在图3的例子中,循环时间可以缩短到10.5分钟。 下列等度泵可以用做纯化系统的柱再生: ( ·Agilent 1100系列单泵(最大流速 10 mL/min, 200 bar ) , 以及 ) Agilent 1100系列制备泵(最大流速100 mL/min 400 bar) 图1 设置阀的窗口 使用梯度泵作为再生泵时,下一步纯化在第二根色谱柱上开始后,可以进行“柱清洗”和“柱平衡”步骤(图4)。然后将冲洗时间直接加到梯度完成之后。 下列梯度泵可以在纯化系统中用于柱再生: ( ·Agilent 1100系列单泵(最大流速 10 mL/min, 200 bar) 含Agilent 1100系列12-位/13-通阀溶剂选择阀 ) ( · Agilent1100系列四元泵(最大 流速10 mL/min, 200 bar) ) 用梯度泵和重叠进样进行交替柱再生 使用具有重叠进样功能的Agilent1100系列制备型自动进样器,可以在前一个纯化运行正在进行的同时,吸取下一个样品。特别是在样品体积很大的制备液相色谱中,可能需要多次吸取进样,这一功能将极大地节省时间(图5)。 图2 运行序列 结论 通过外接Agilent 1100系列2-位/10-通阀,使用交替柱再生,可以进行高通量纯化分离。在第二根柱上的纯化分离开始后,可以进行柱清洗,或柱清洗和柱平衡(取决于再生泵的类型)。此外,用自动进样器累加进样,缩短了周期时间,特别是制备液相色谱,一般都是进样体积大、进样时间长。 图3 用等度再生泵进行交替柱再生 图4 用梯度再生泵进行交替柱再生 图5 用梯度再生泵和重叠进样进行交替柱再生 ( ' Preparative High Throughput HPLC-Alternating Column Regeneration with the Agilent 1100 Series Valve Solutions (制备型高通量HPLC-用Agilen t 1100系列阀解决方案进行交替柱再生),发行号5988-8085EN。 ) 应用 技术 纯化策略 阀的应用 两个应用实例说明了Agilent 110012-位/13-通阀在纯化系统中的应用。在第一个例子中,将其用于回收收集,样品的剩余部分不是作为馏分,而是收集到一个专门的容器里。如果在纯化分离中出现错误,例如,由于参数设置不当,样品没有流入废液,1,而是存在一个容器中,很容易回收。第二个例子是将阀用于基于时间的馏分收集。 结果与讨论 化学工作站软件提供了回收收集功能。即,所有不能作为馏分收集的成分都进入一个专门的容器,很容易回收(图1)。回收收集需要使用带漏斗盘(G1364-84502)的Agilent1100系列馏分收集器AS。1100系列馏分收集器PS的针头太短,不能使用漏斗。 回收收集 回收收集也可以通过设置12-位/13-通阀进入馏分收集器废液管完成,两种馏分收集器AS或PS都可以使用。 图1回收收集 回收收集必须检查Next positionafter run框。自动将Position设在Use current。用这种置置,当一次纯化完成后,阀自动切换到下一个回收位置,在下一次运行开始时,保留这个位置。即,样品1收集到位置1,样品2收集到位置2,以此类推。先决条件是,在第一次运行开始前,将阀手动切换到位置1。用12-位/13-通阀最多可以收集12个样品。如果需要更多的回收位置,在第一阀出口端连接更多的12-位/13-通阀。唯一的限制是,可以连接到系统上的阀的最大数目和为每个连接阀所设置的更复杂的方法。因为这次分离完成后及下一次分离后运行之前,阀要切换到下一个位置,建议方法包含后运行时间,例如,把柱平衡计入运行时间。因此,在色谱柱清洗和平衡期间,来自色谱柱的流动相仍然被收集到样品回收位置(不属于下一个样品的回收位置)。方法实例见表1a和1b。 基于时间的馏分收集 Agilent 1100系列12-位/13-通阀也可以用于简单的馏分收集,连接在检测器上,而不是Agilent 1100系列留分收集器。馏分只能通过阀切换用时间表按时间段收集(图3)。最后的位置用做废液位置,!收集不分析时的流动相。由于固定的位置作为方法的起始位置,在分离完成后阀将切换回到这个位置。因此,在平衡、进样过程中从色谱柱中出来的流动相,将进入废液位置,不会稀释或污染任何馏分。当使用按顺序设置这些阀的方法时,,可以进行合并,甚至从不同的样品瓶中进样。用Agilent 1100系列12-位/13-通阀基于时间进行馏分收集,只能使用化学工作站软件-不需要,也不支持纯化/高通量软件。 结论 1100系列12-位/13-通阀和1100系列纯化系统联用,可以用于: ·连接到馏分收集器废液管路,进行回收收集 。在简单的纯化系统中,代替馏分收集器进行基于时间的馏分收集 图2 设置阀的窗口 含后运行时间 梯度开始: 0分钟10%B 9分钟90%B 10分钟90%B 停止时间: 10分钟 后运行时间: 3分钟 表1a 包含平衡色谱柱的后运行时间的梯度分离条件 不包含后运行时间 梯度开始: 0分钟10%B 9分钟90%B 10分钟90%B 10.1分钟10%B 13分钟10%B 停止时间: 13分钟 后运行时间: 关闭 表1b 图3 平衡包含在运行时间内 用12-位/13-通阀进行的基于时间的馏分收集 ( Recovery Collection a nd Time-based Fraction Collection-Preparative HPLC with the Agilent 1100 Series Valve Solutions s” (回收收集和基于时间的馏分收集-使用Agilent 1100系列阀解决方案的制备液相色谱),谱行号5988-8225EN。 ) 备注 备注 安捷伦科技公司生命科学与化学分析部客 户服务中心免费专线:800-820-3278 www.agilent.com/purification C安捷伦科技有限公司。2007 2007年11月中国印刷发行号5989-5948CHCN Agilent Technologies 问我们的网站: www.agilent.com/chem/purification,,下载应用报告全文。 Agilent 1200系列纯化系统是药物高通量合成小分子和大分子化合物库纯化的通用工具,能够得到高纯度馏分,获得目标化合物的高回收率。Agilent 6110/6120系列MSD是新一代单级四极杆仪器,可以进行简便而可靠的质谱引导的馏分收集,也可以完成更复杂的纯化工作,如组合离子源或使用正/负极切换等。本应用报告报道了用不同电离技术,如ESI和APCI,以正电离和负电离模式,用Agilent 6110/6120 MSD进行质谱引导的馏分收集。可以用Agilent 6110 MSD ESI或APCI源,正或负离子模式,质谱引导的进行简便可靠的馏分收集。如果想要在一次运行中使用组合离子源,同时进行ESI 和APCI, 就需要使用Agilent 6120 MSD。
确定











































还剩74页未读,是否继续阅读?

产品配置单




安捷伦科技(中国)有限公司为您提供《药物高通量合成小分子和大分子化合物中馏分收集与纯化检测方案(液相色谱仪)》,该方案主要用于其他中其他检测,参考标准--,《药物高通量合成小分子和大分子化合物中馏分收集与纯化检测方案(液相色谱仪)》用到的仪器有Agilent 1290 Infinity II 制备型液相色谱、Agilent 6470 三重四极杆液质联用系统、Agilent 1290 Infinity II Multisampler、OpenLAB 软件
推荐专场











相关方案
更多
该厂商其他方案
更多