








方案详情
文
赛默飞世尔科技双三元液相色谱系统,双三元系统是UltiMate® 3000系列色谱的卓越组合,通过共享自动进样器、柱温箱、软件实现两套分析系统的功能。 无论是常规分析、微量分析或纳升级分析,双三元系统均能提供完美的解决方案。 UltiMate® 3000系列色谱仪凭借其卓越的性能、创新的理念、丰富的配置, 在2006年匹兹堡展会上荣获IBO金奖
赛默飞双三元液相色谱-人血浆中苯芴醇检测
方案详情

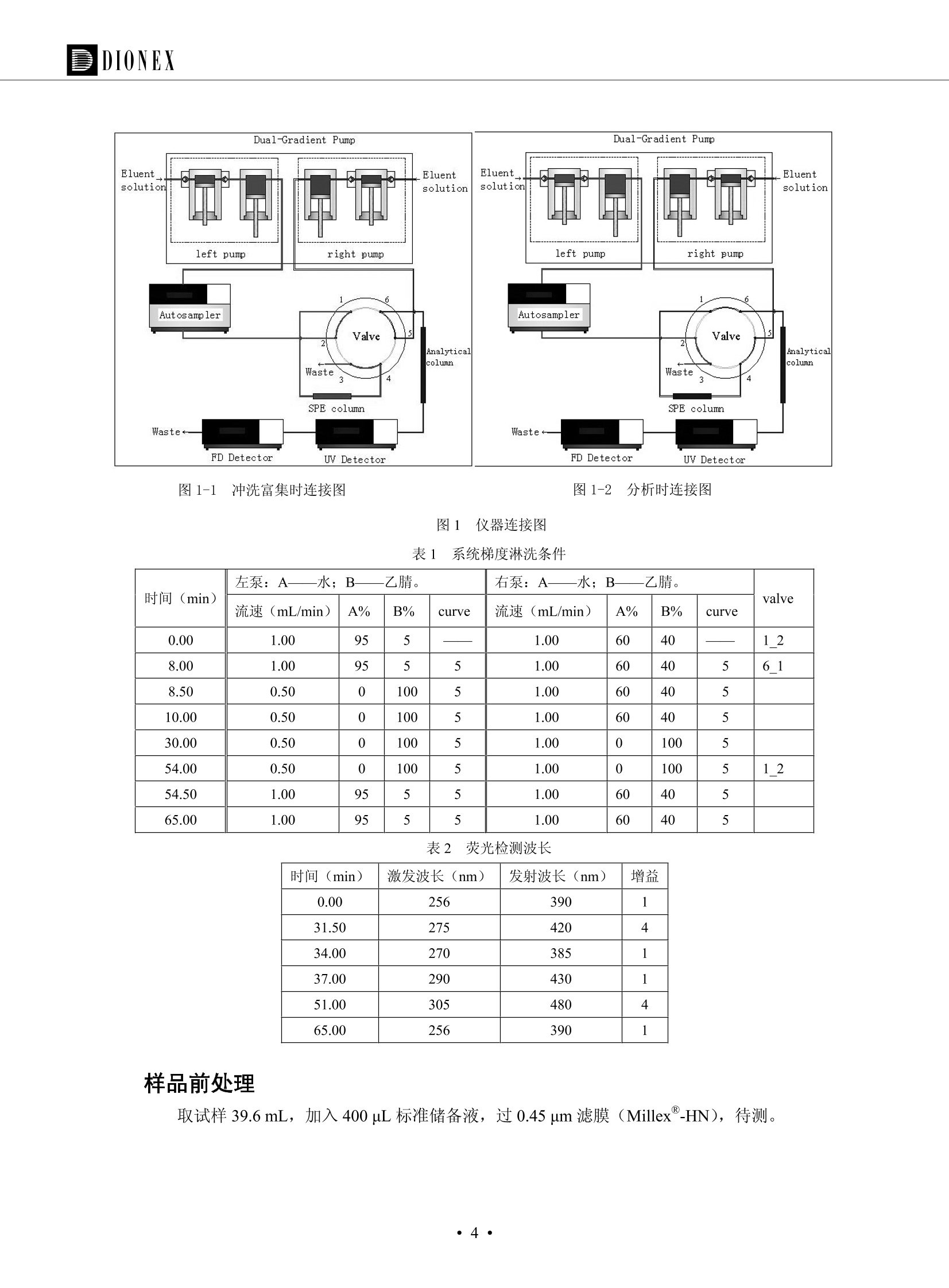
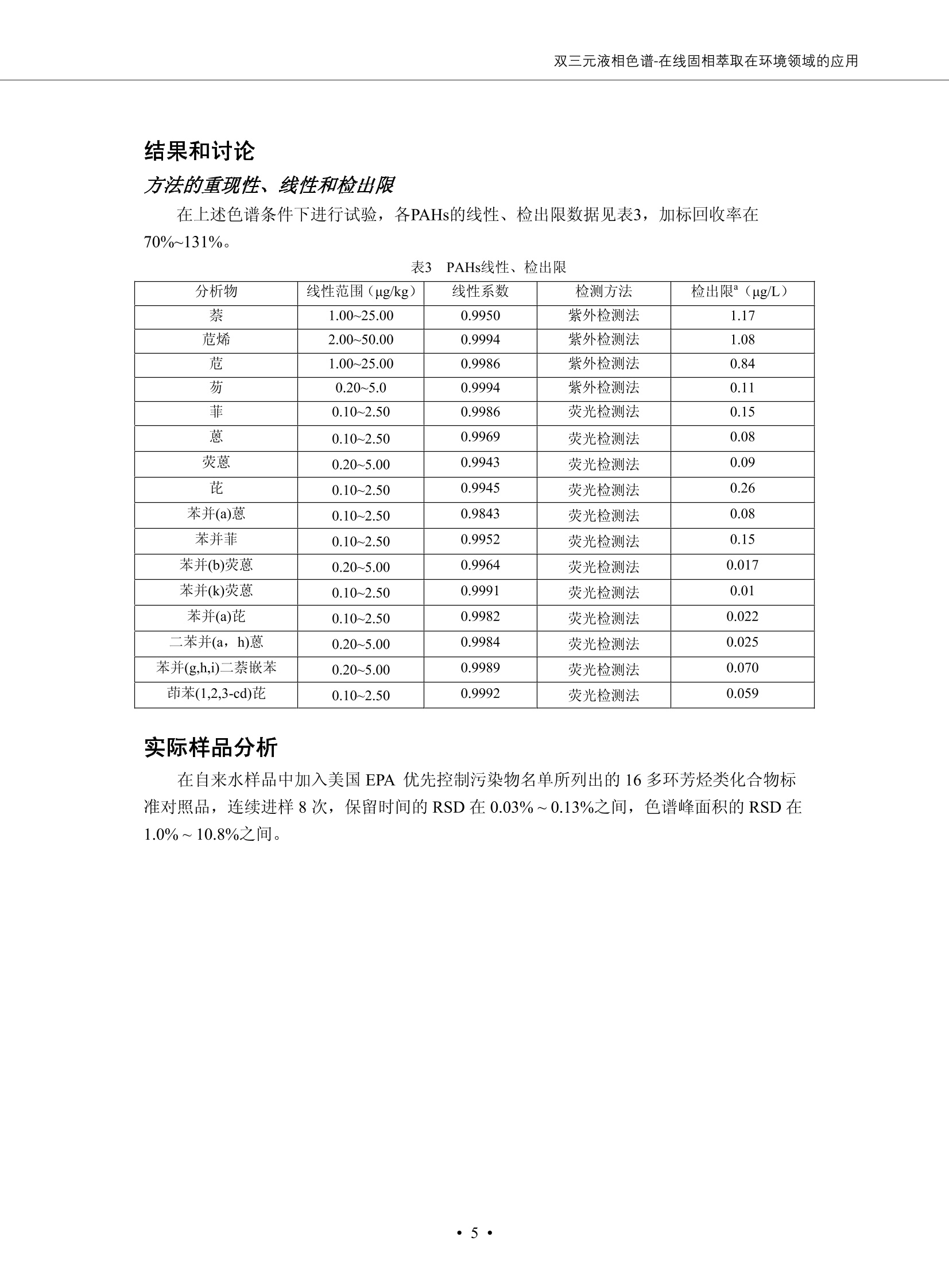
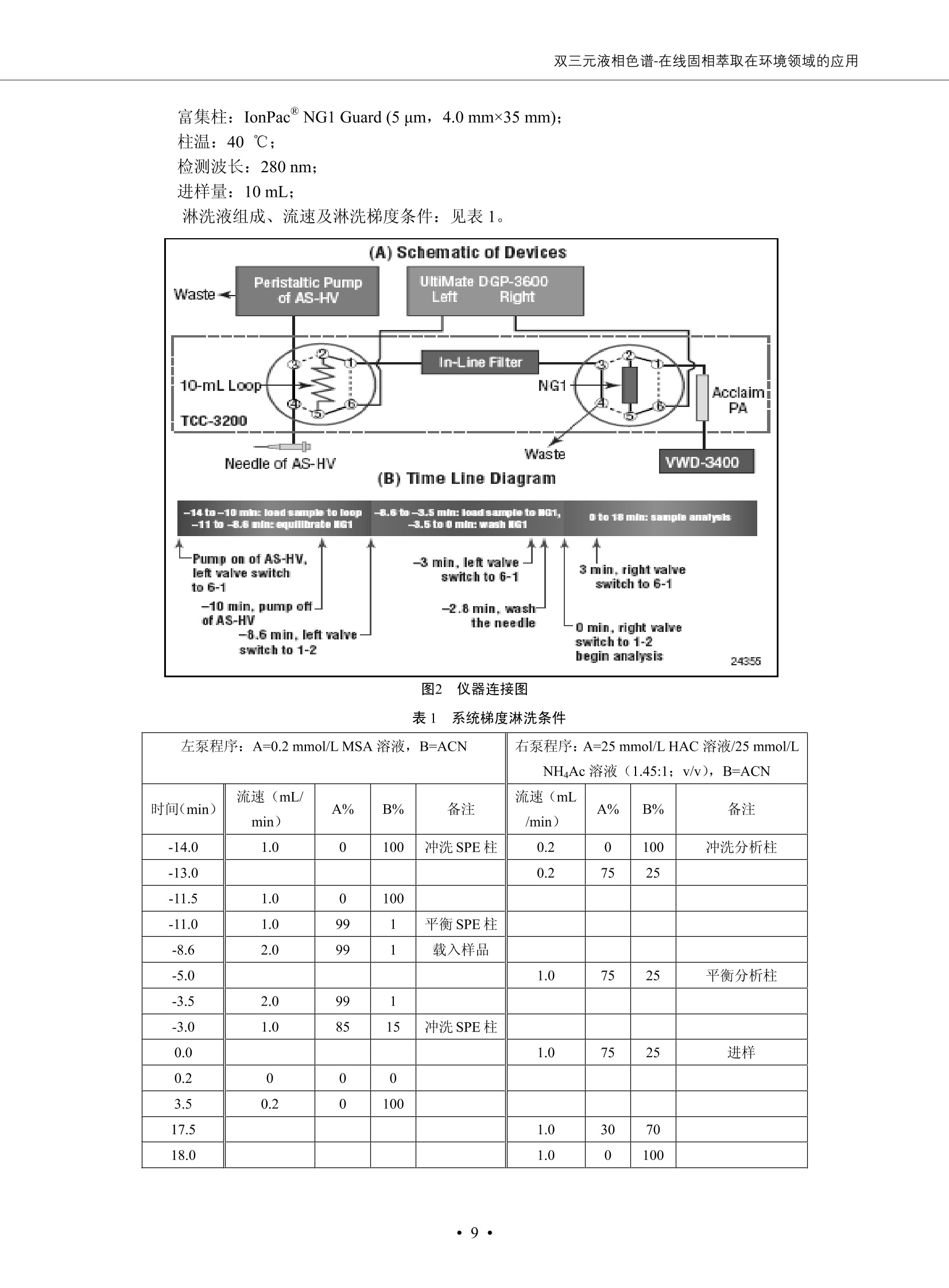
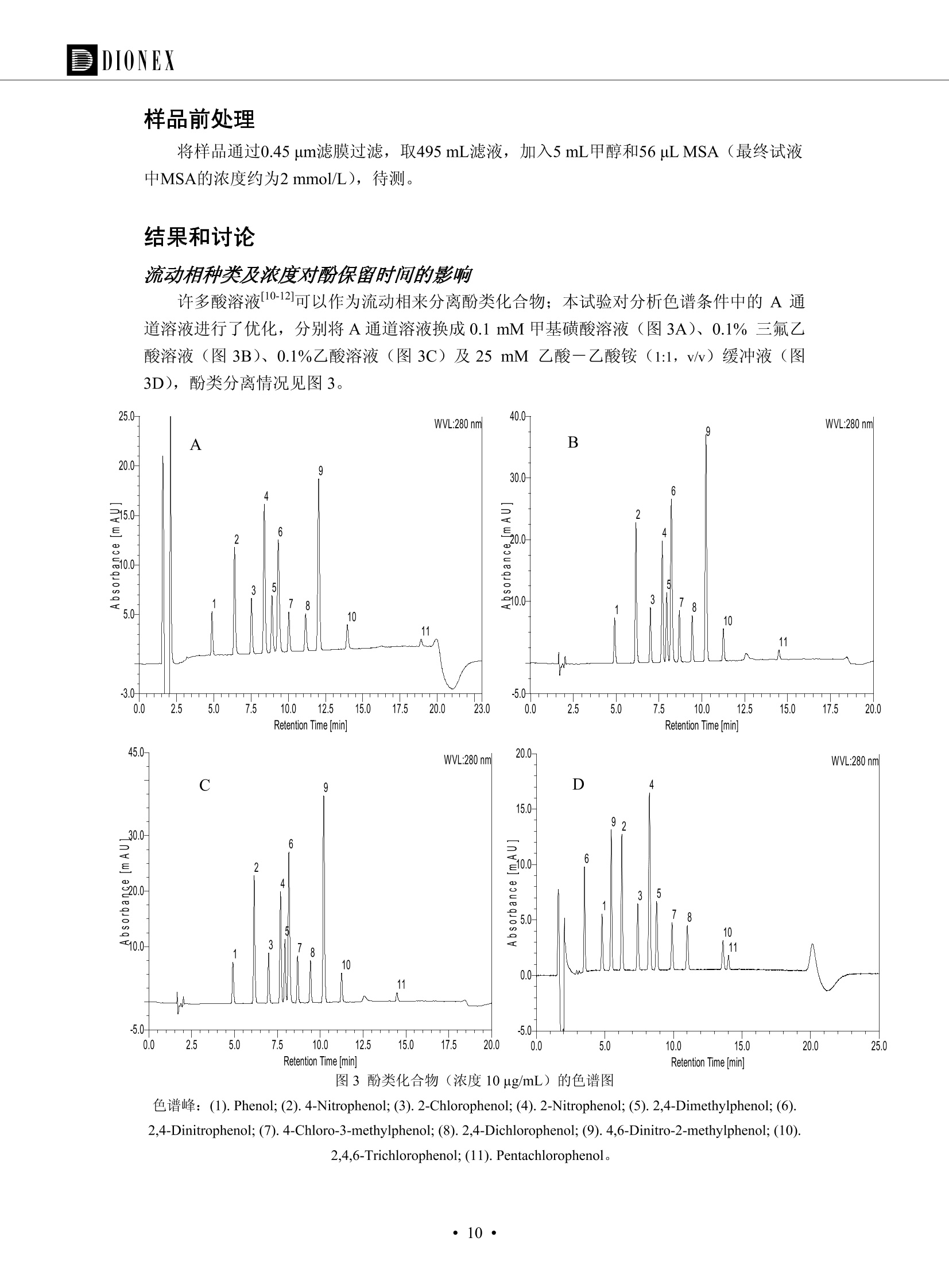
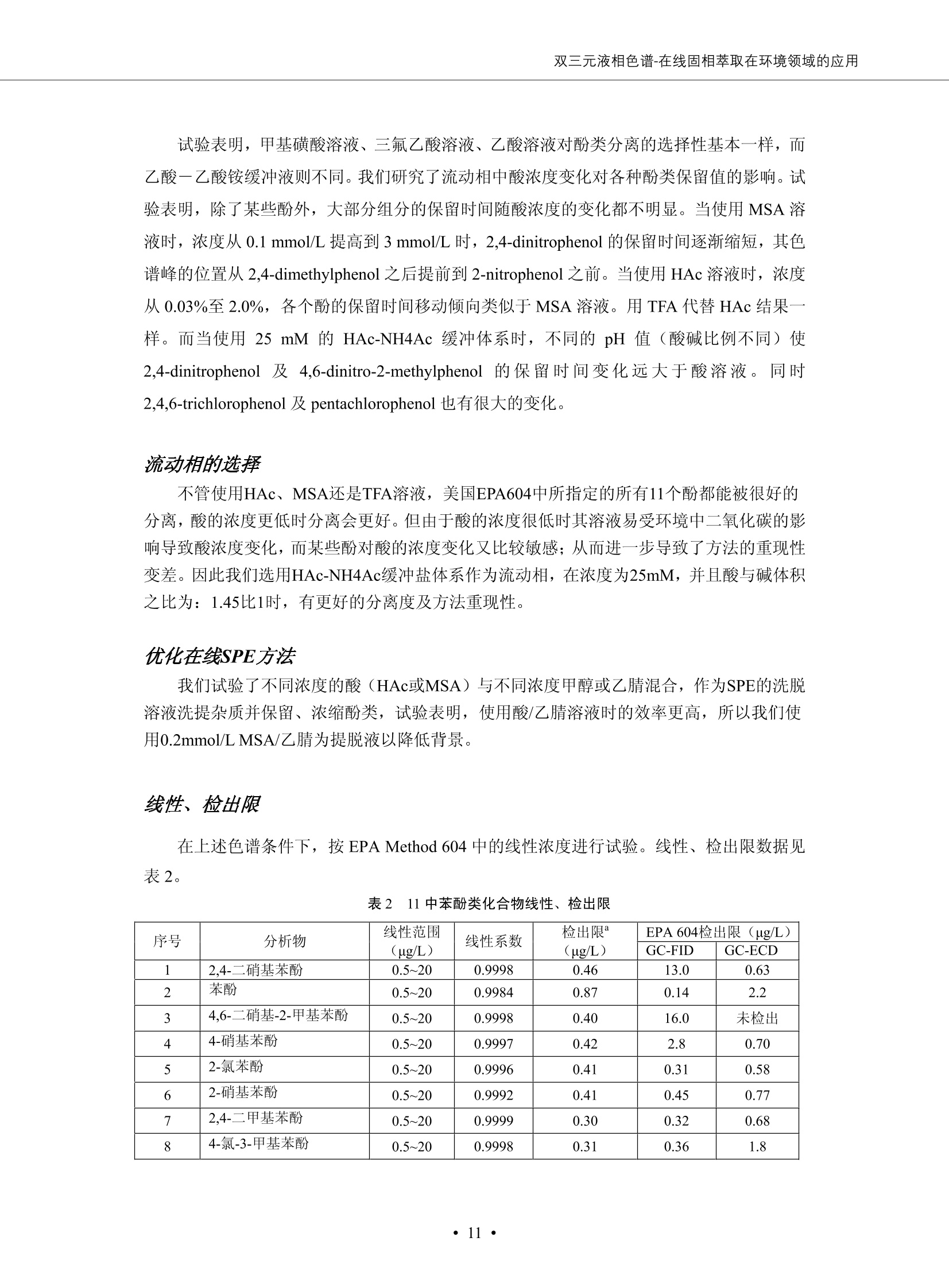
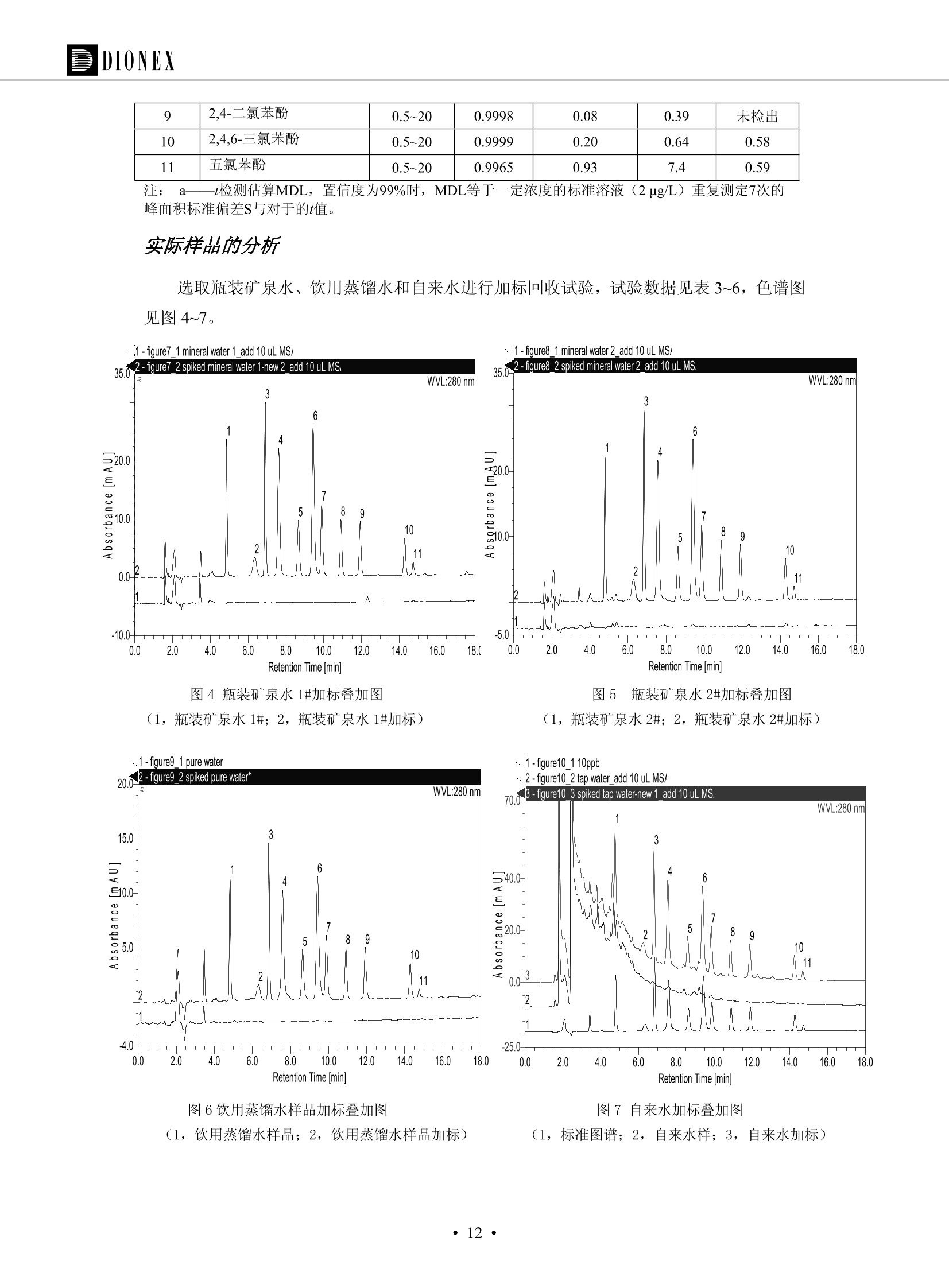
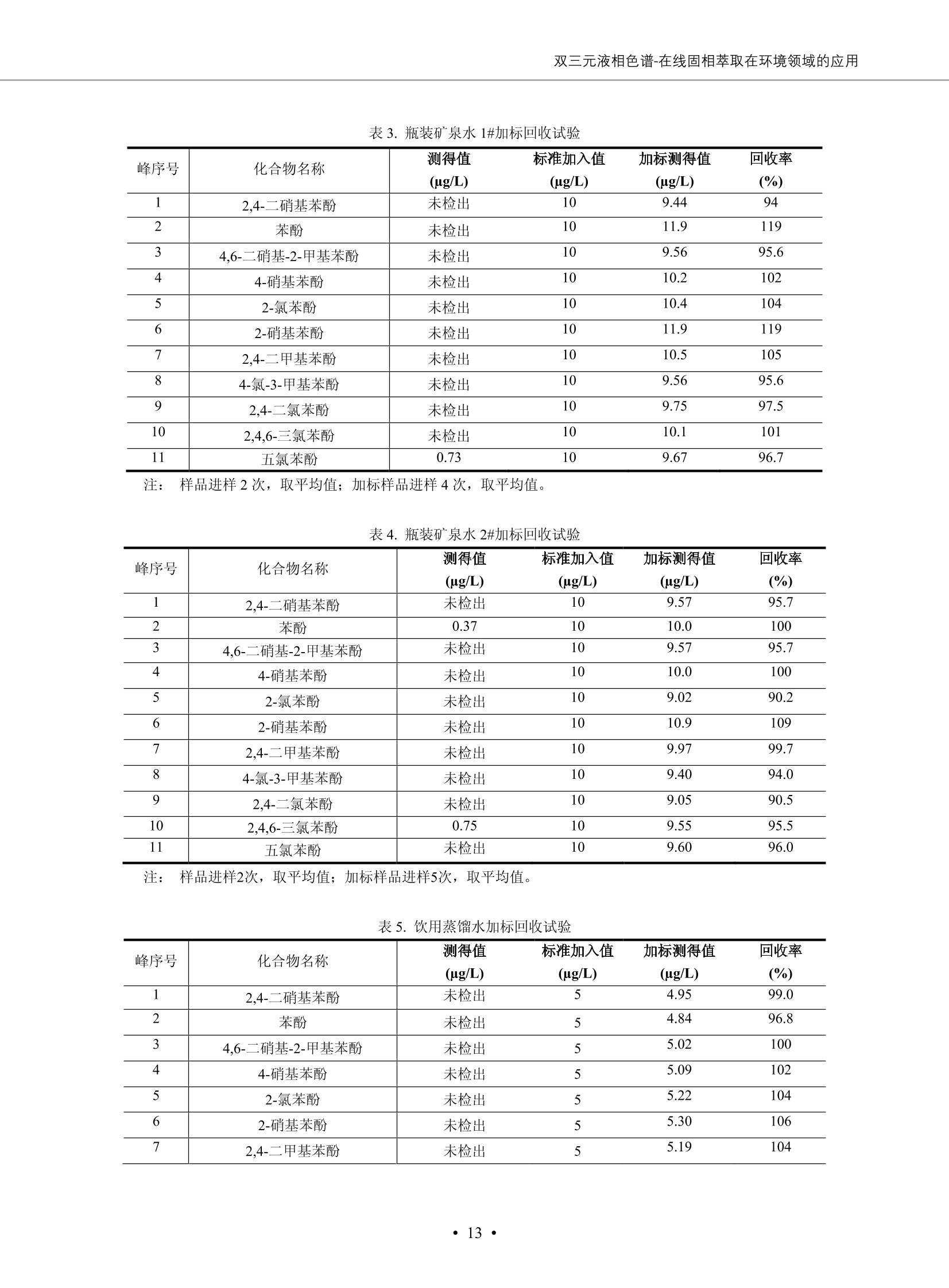
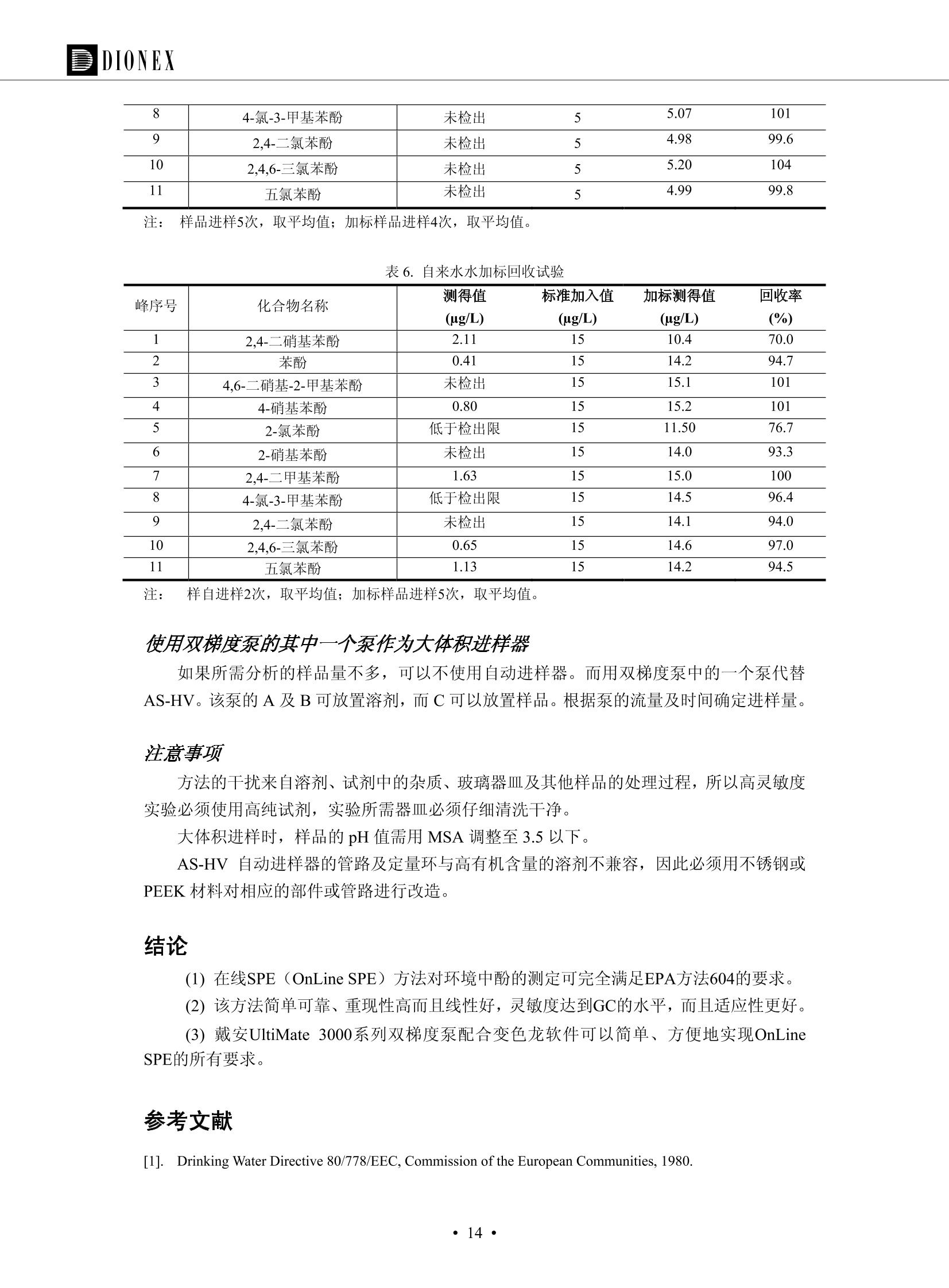
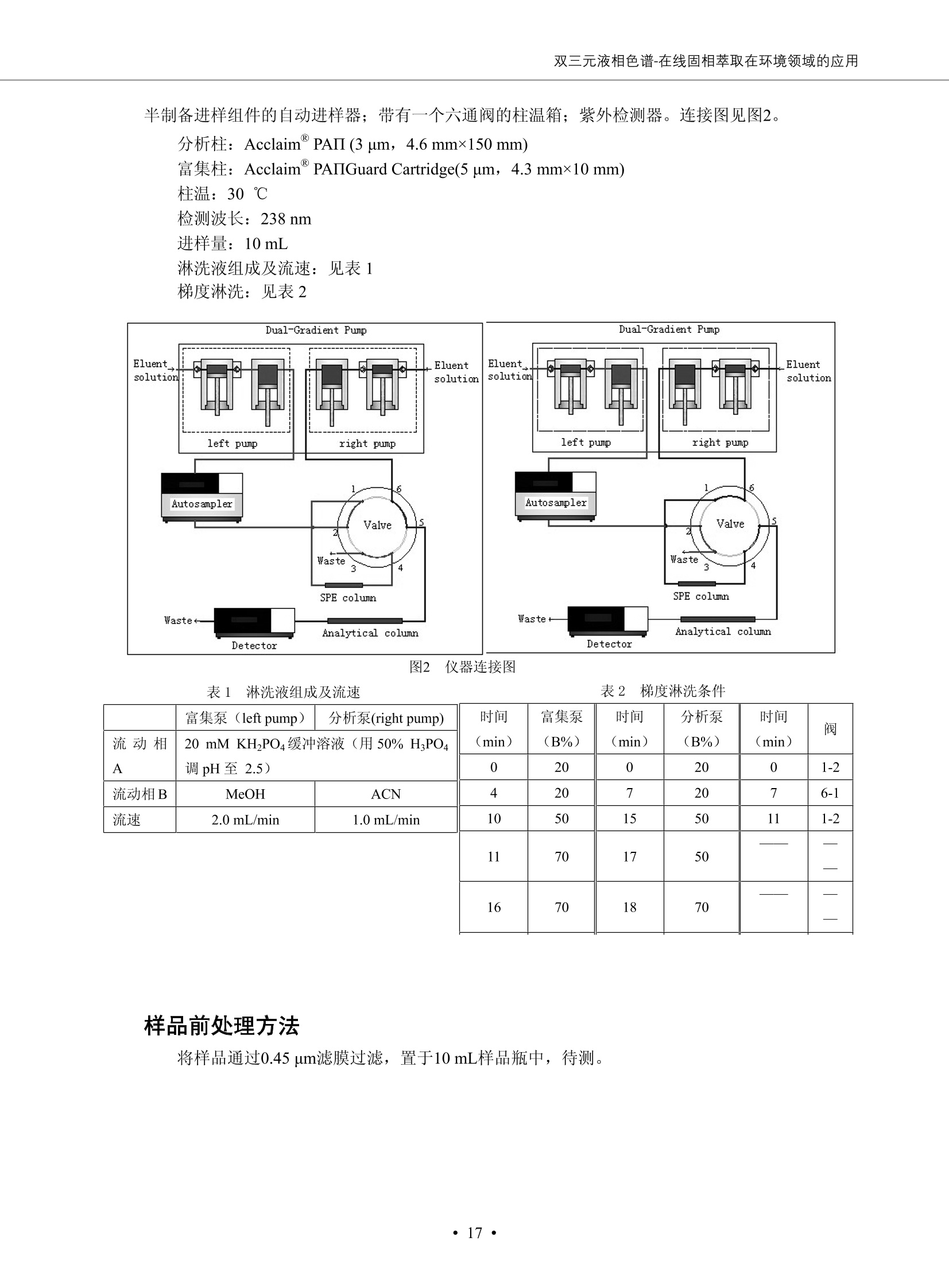
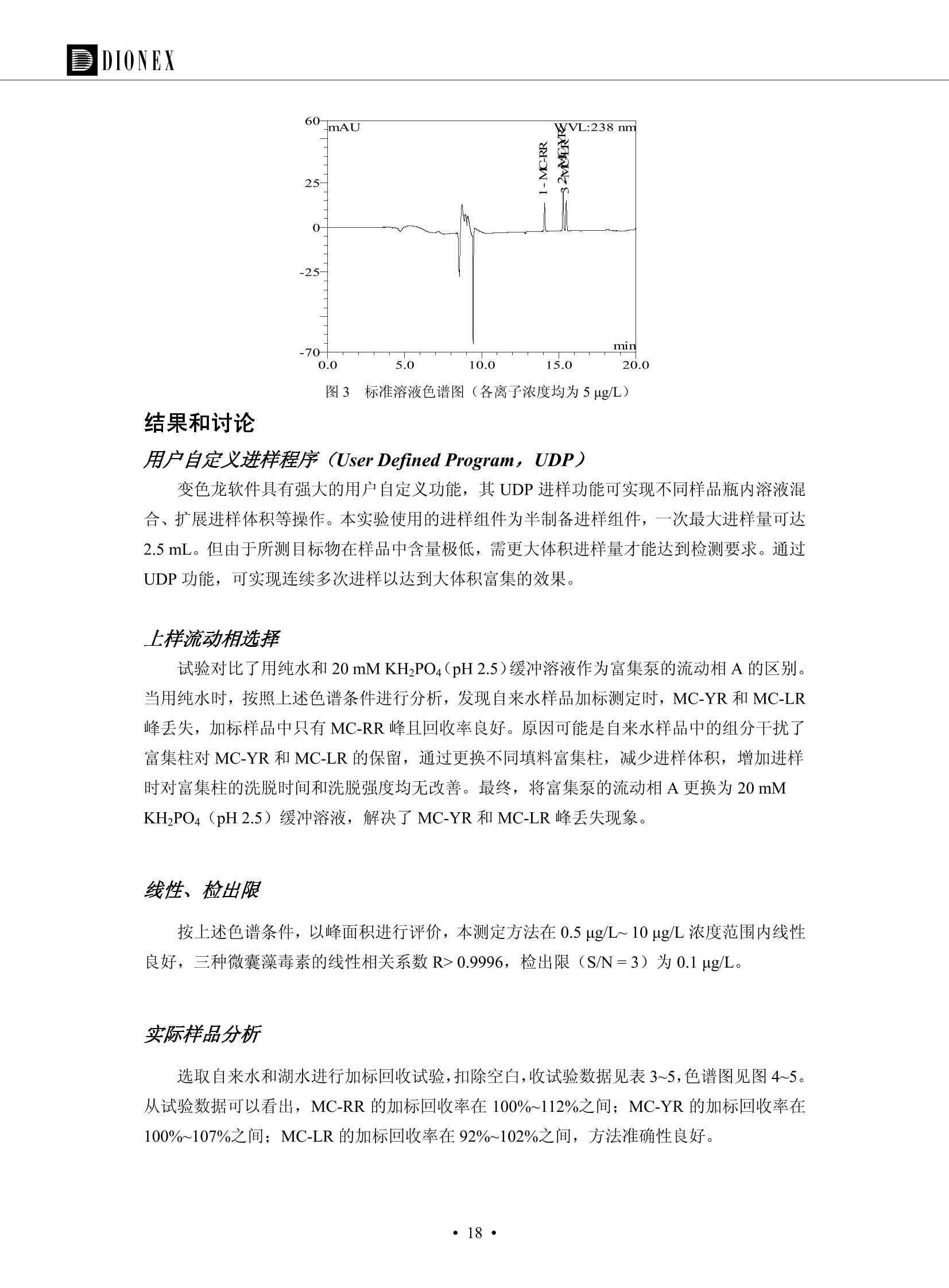
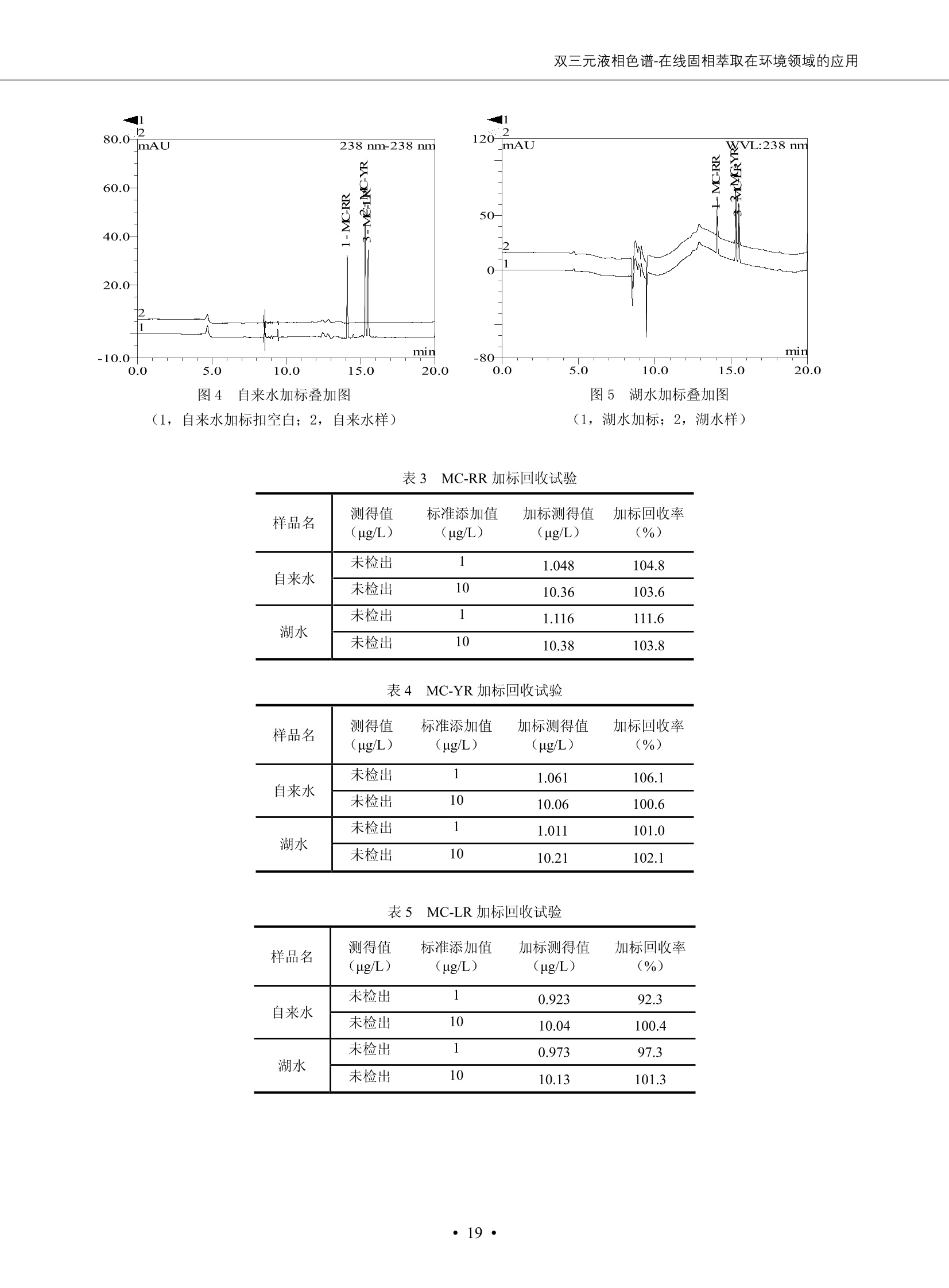
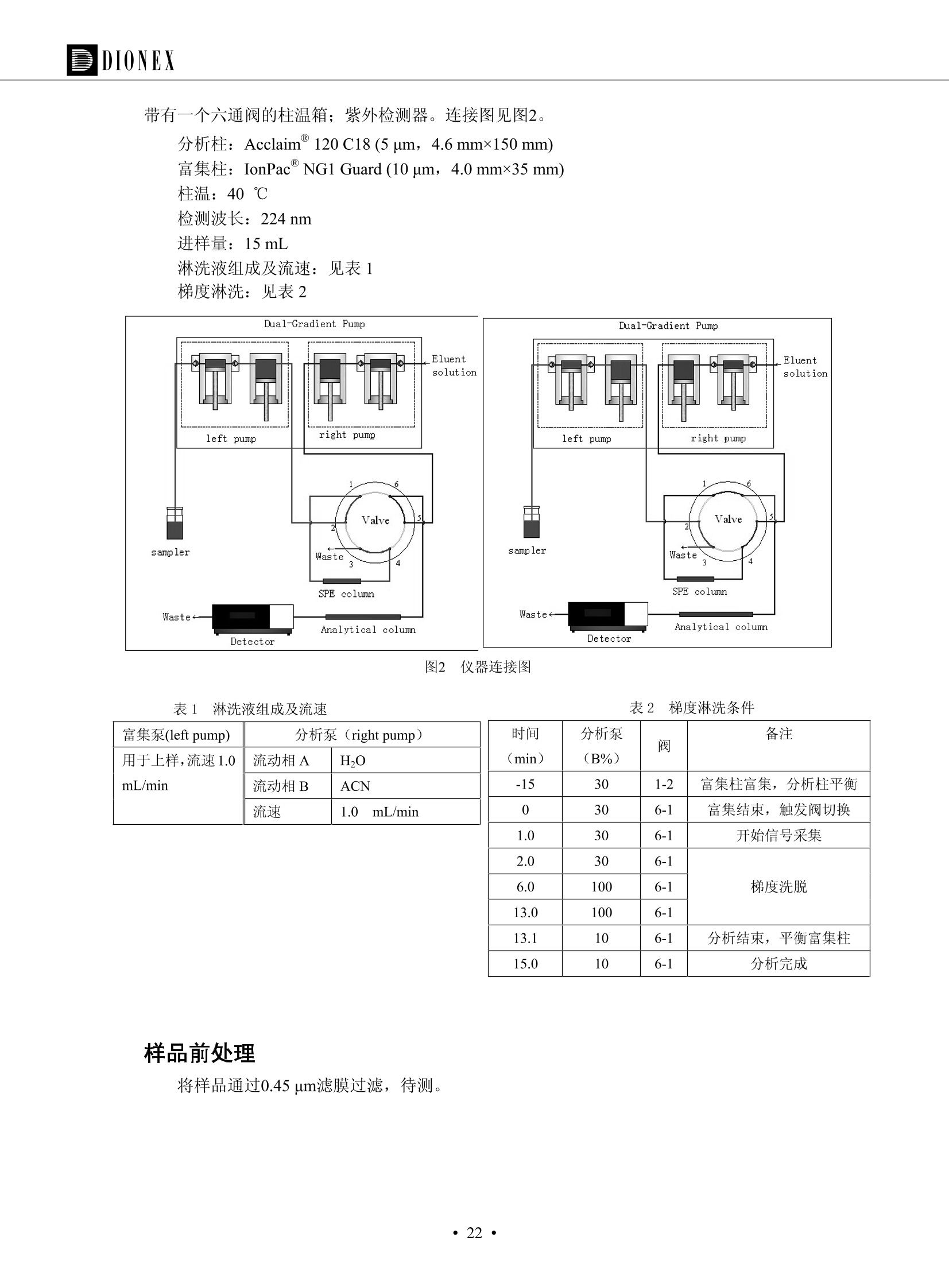
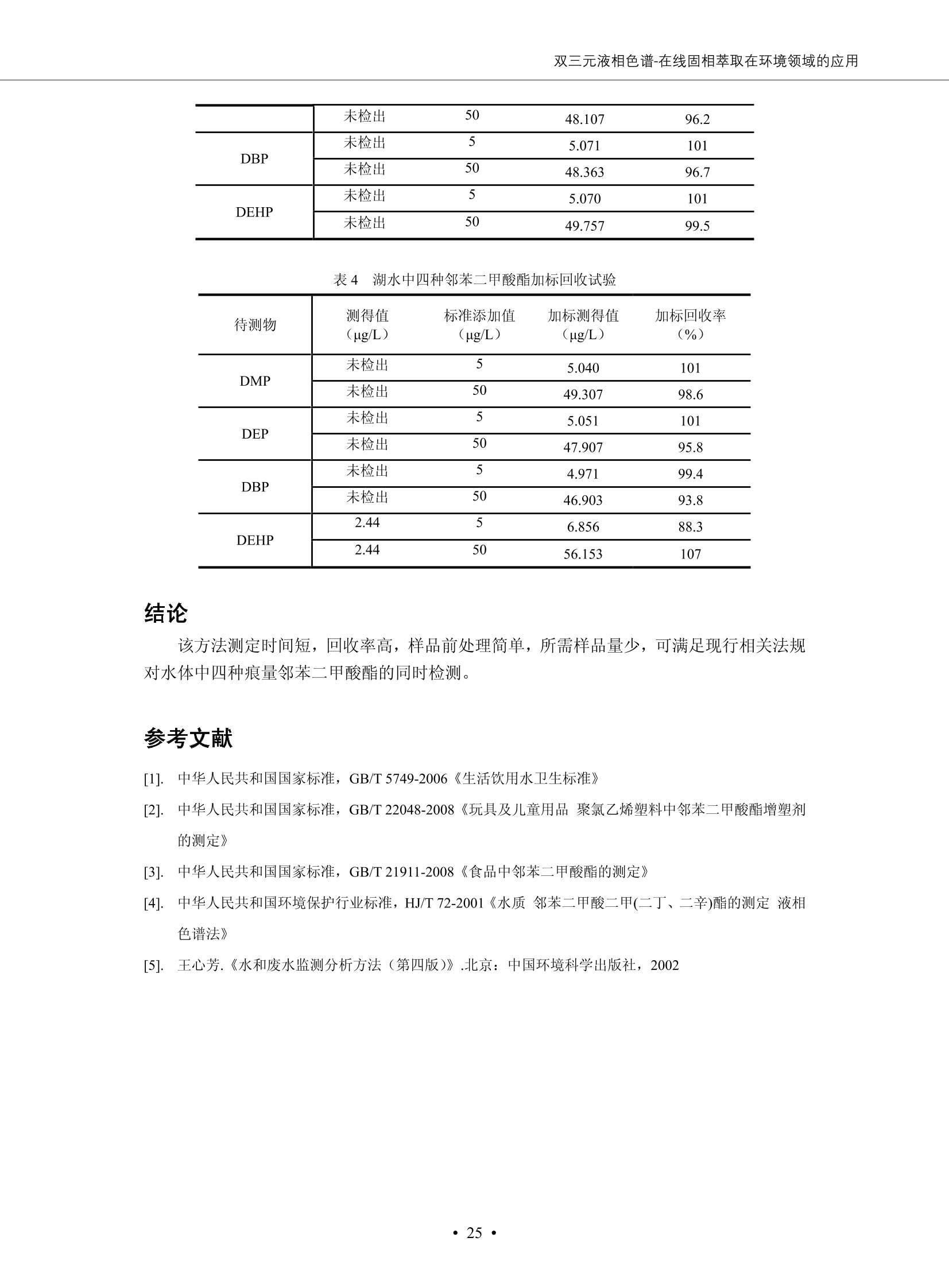
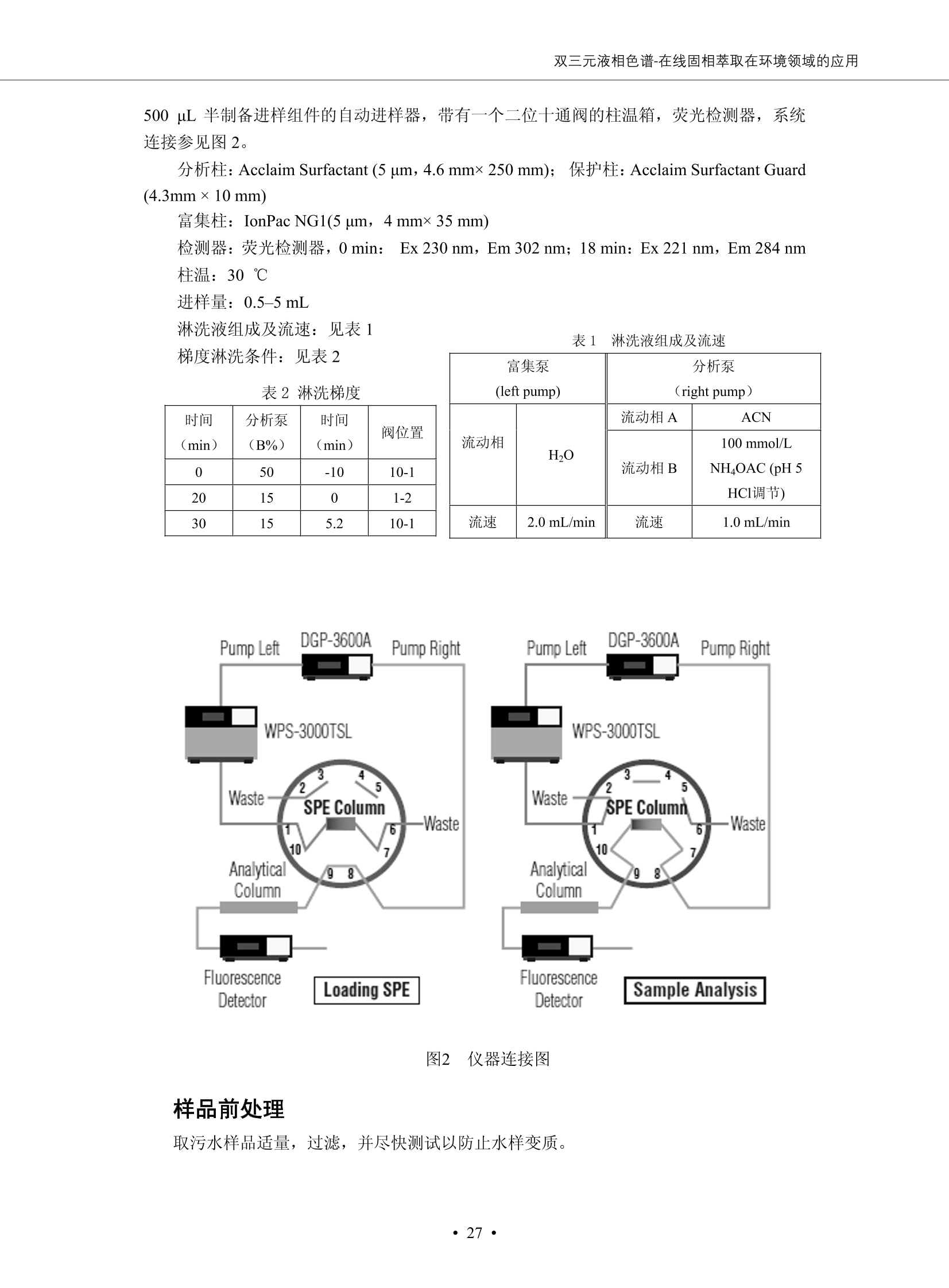
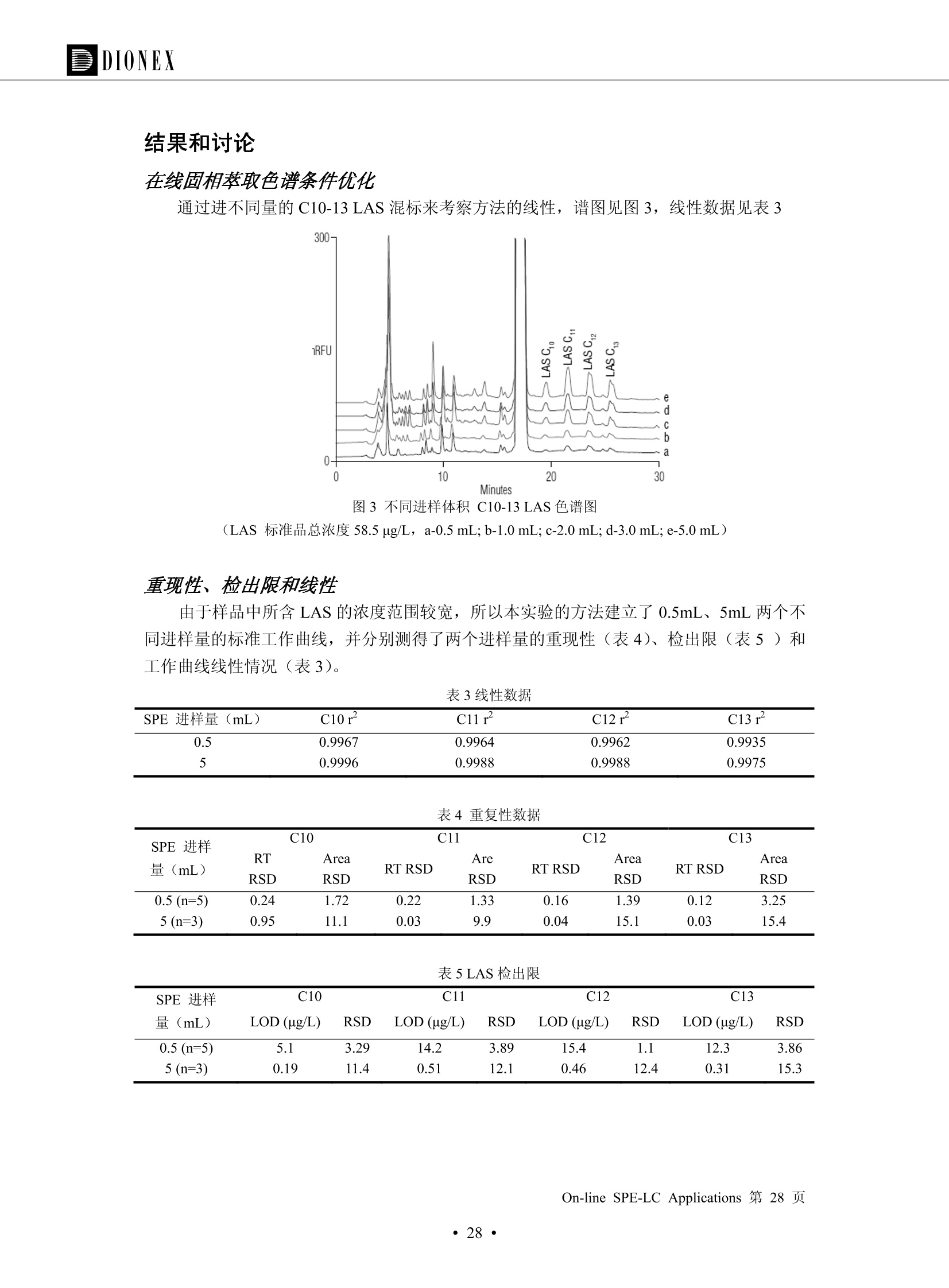
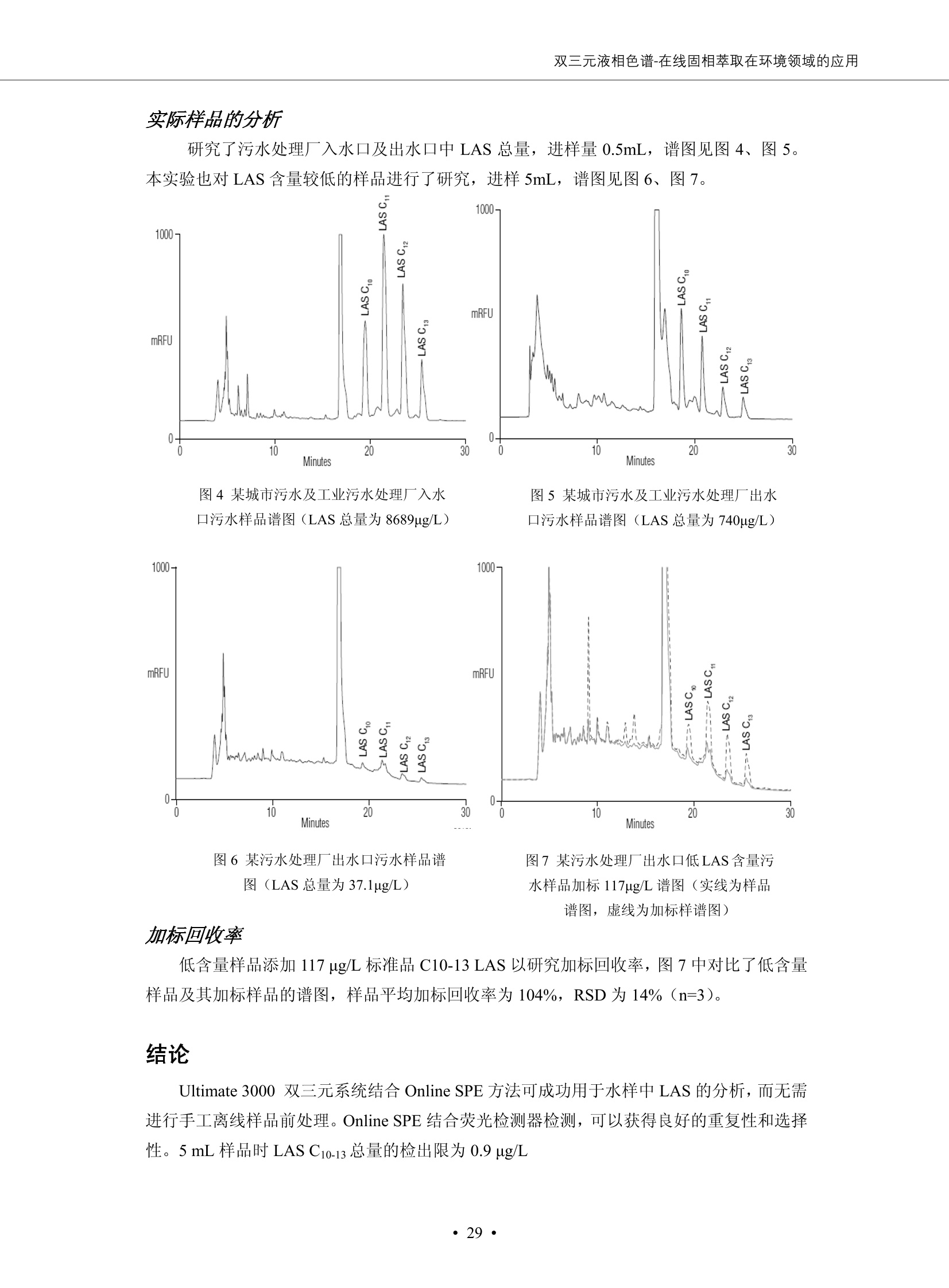
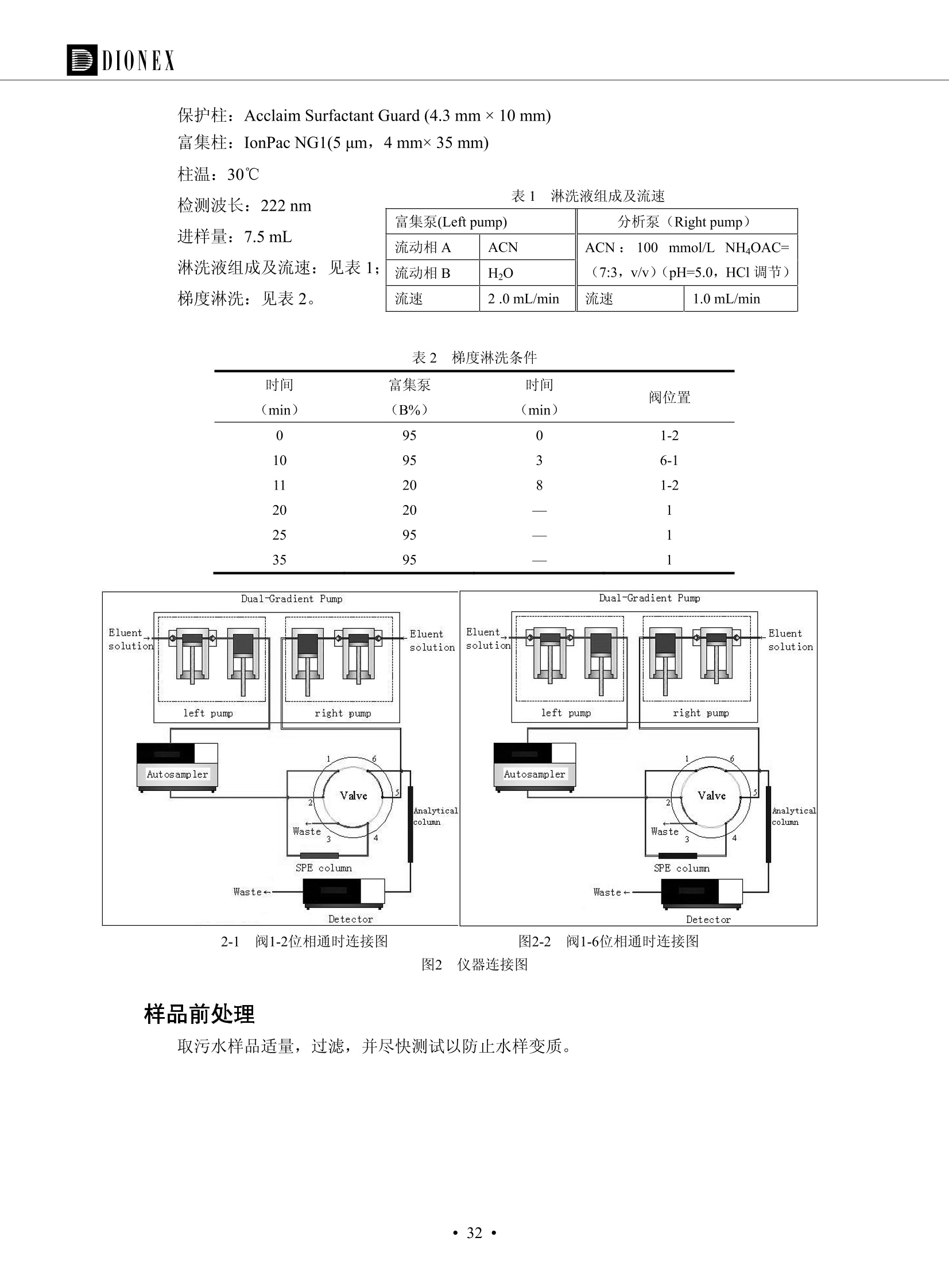
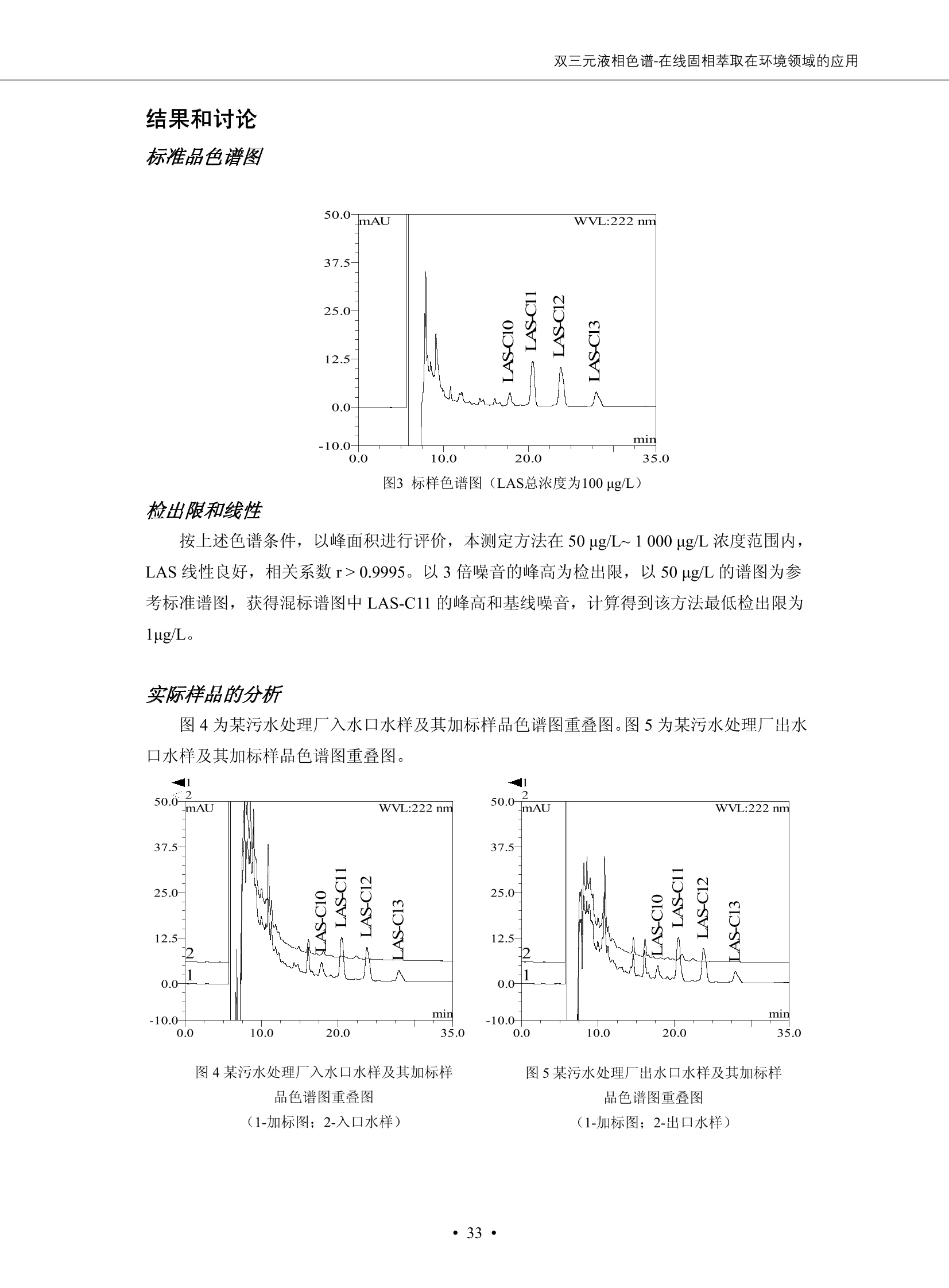
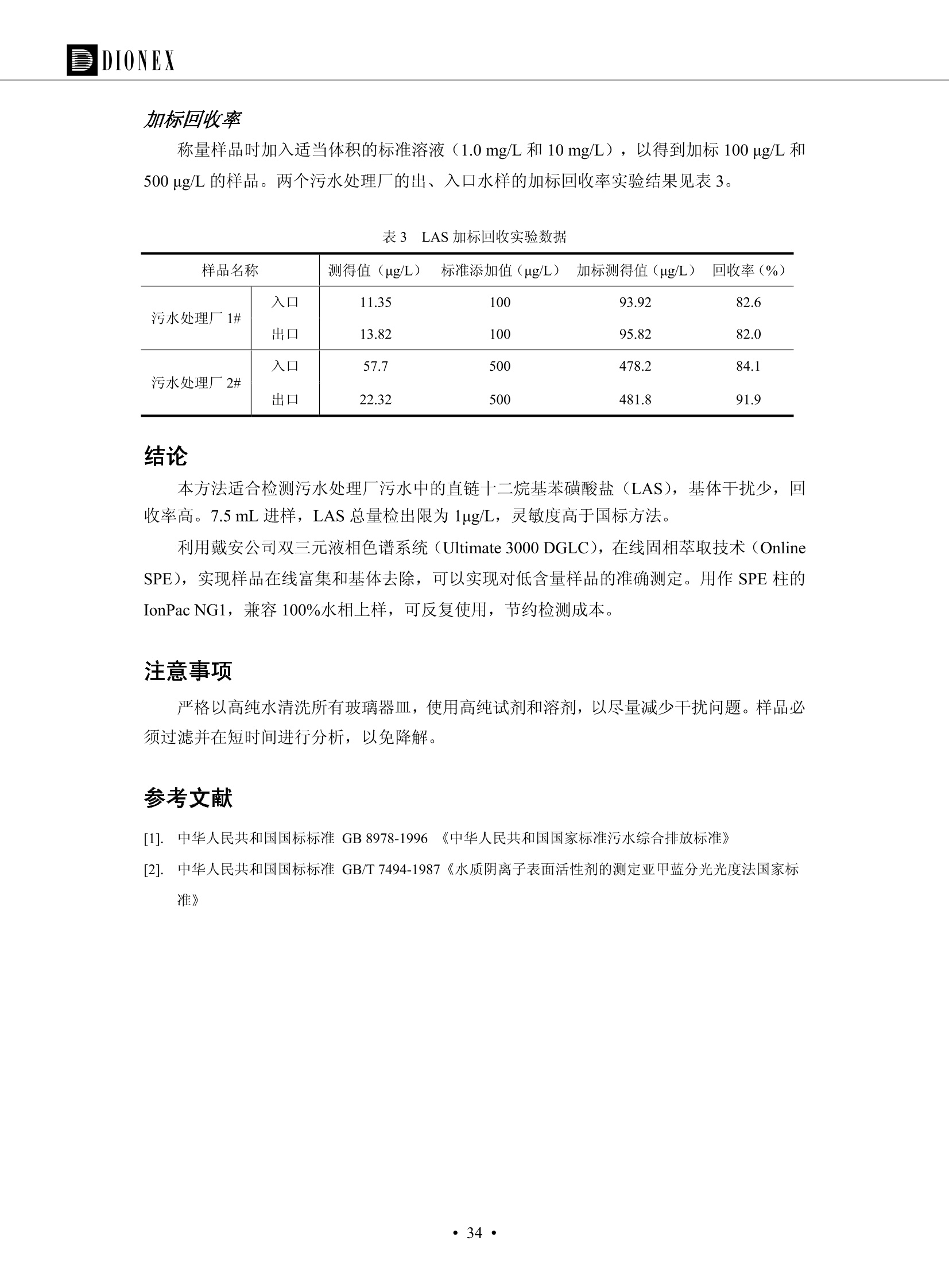
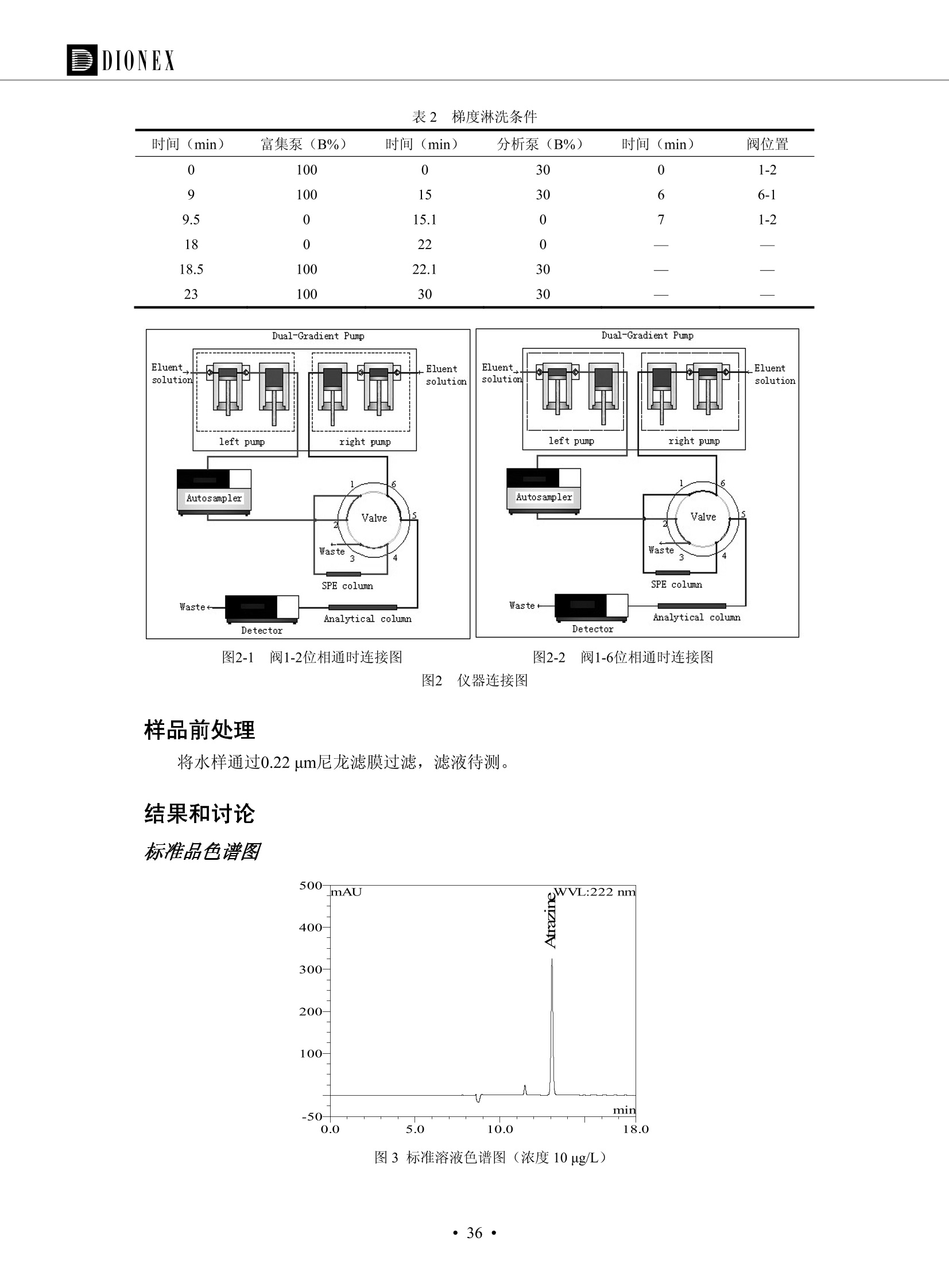
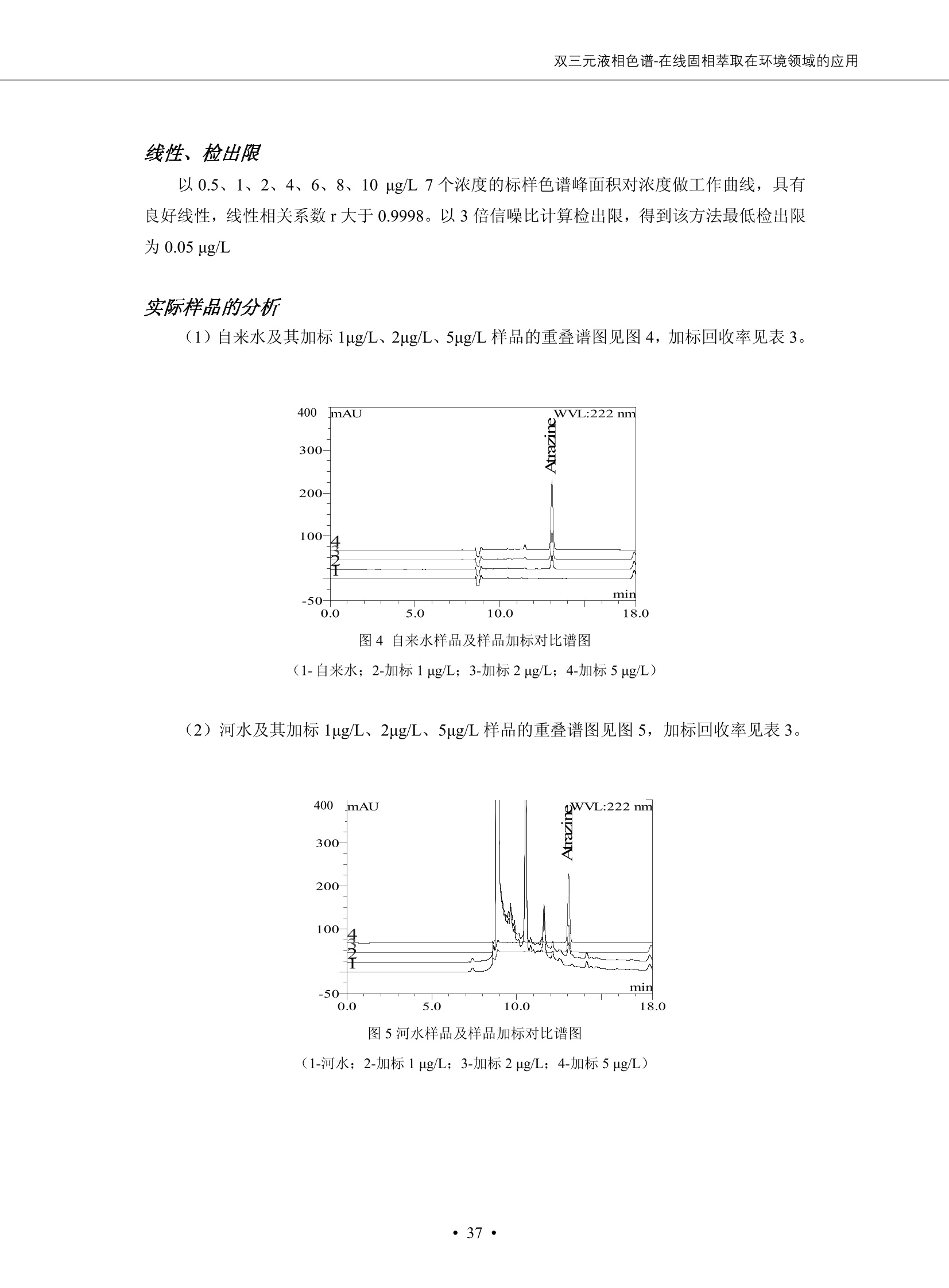
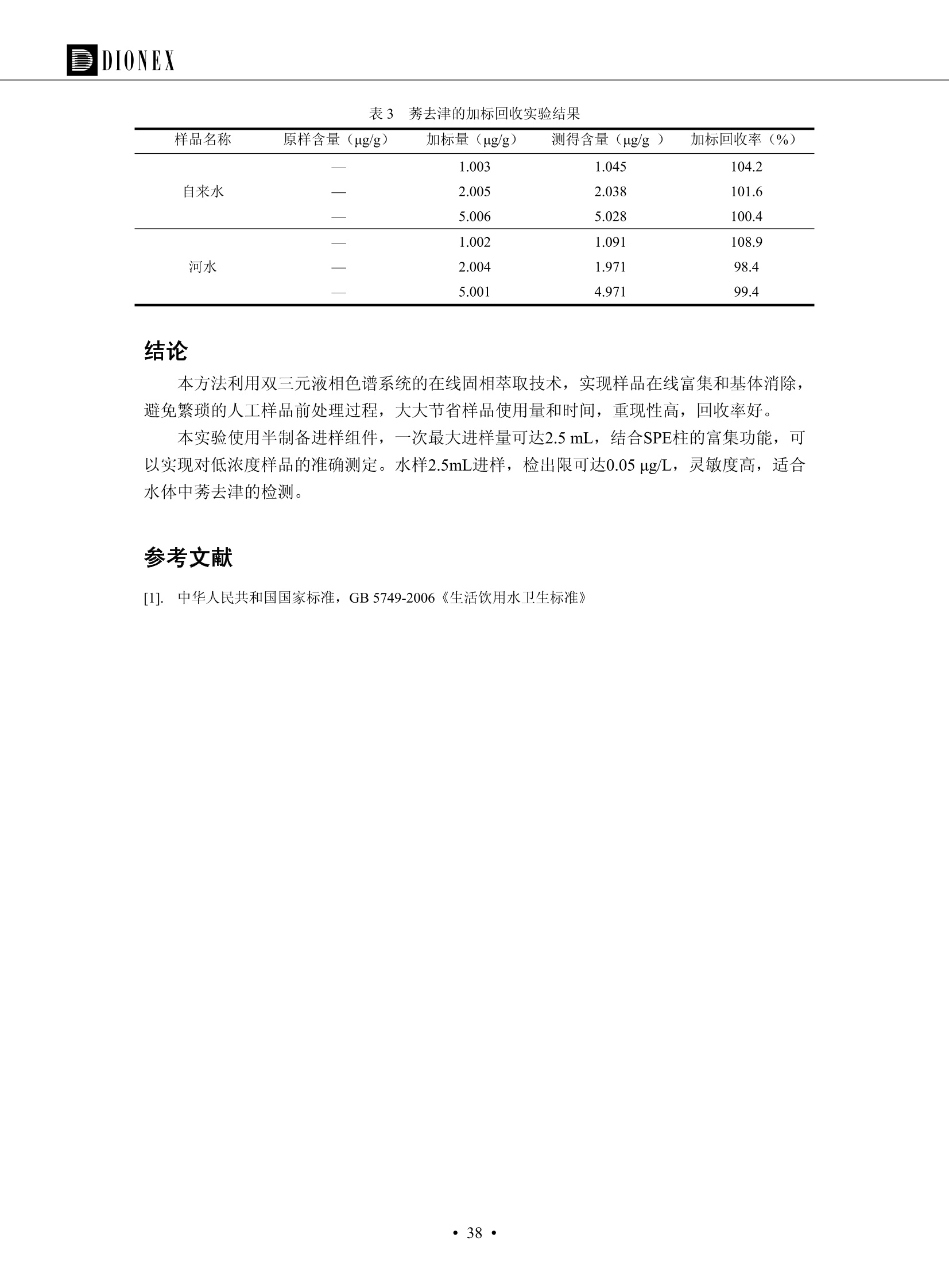
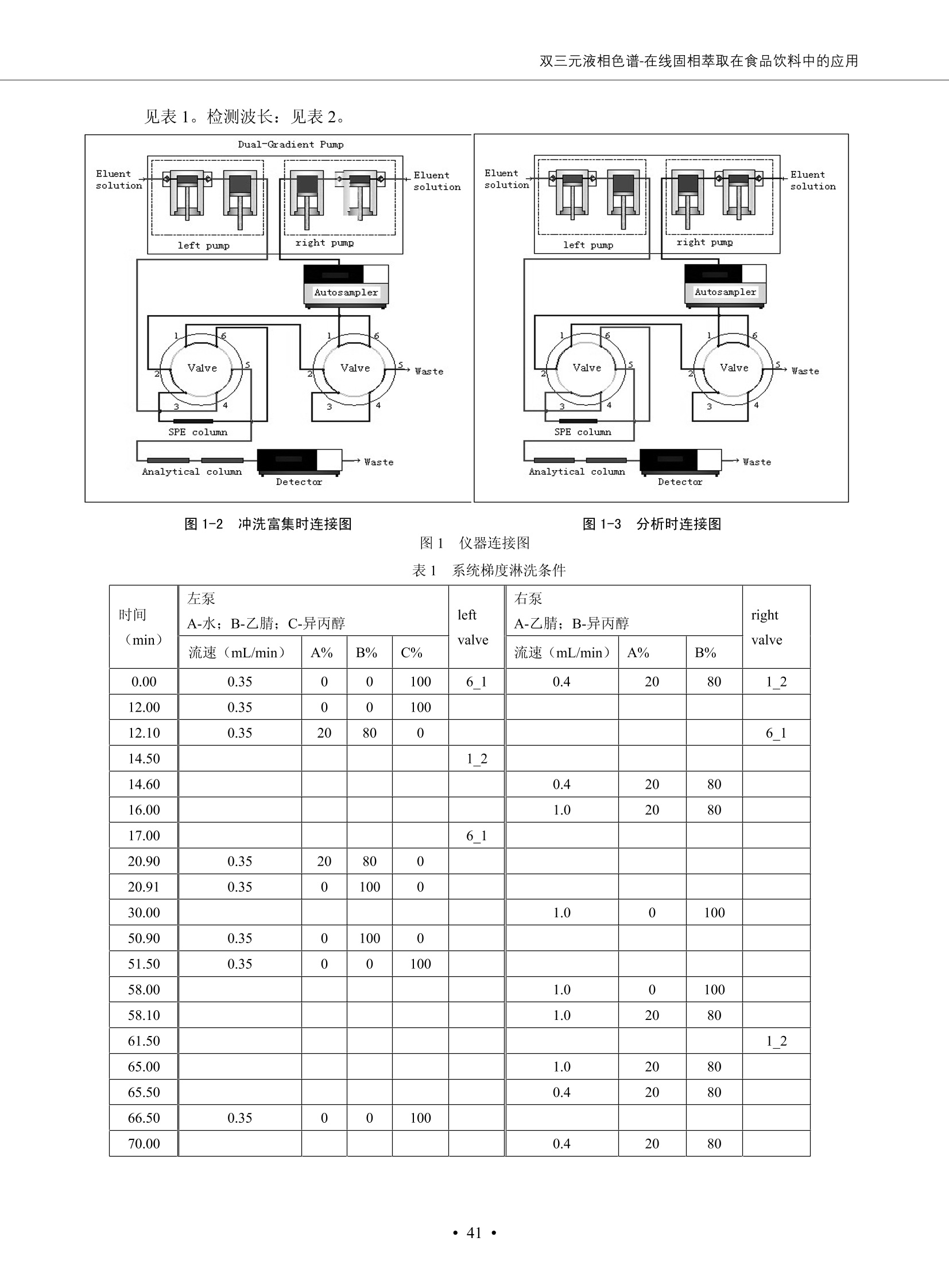
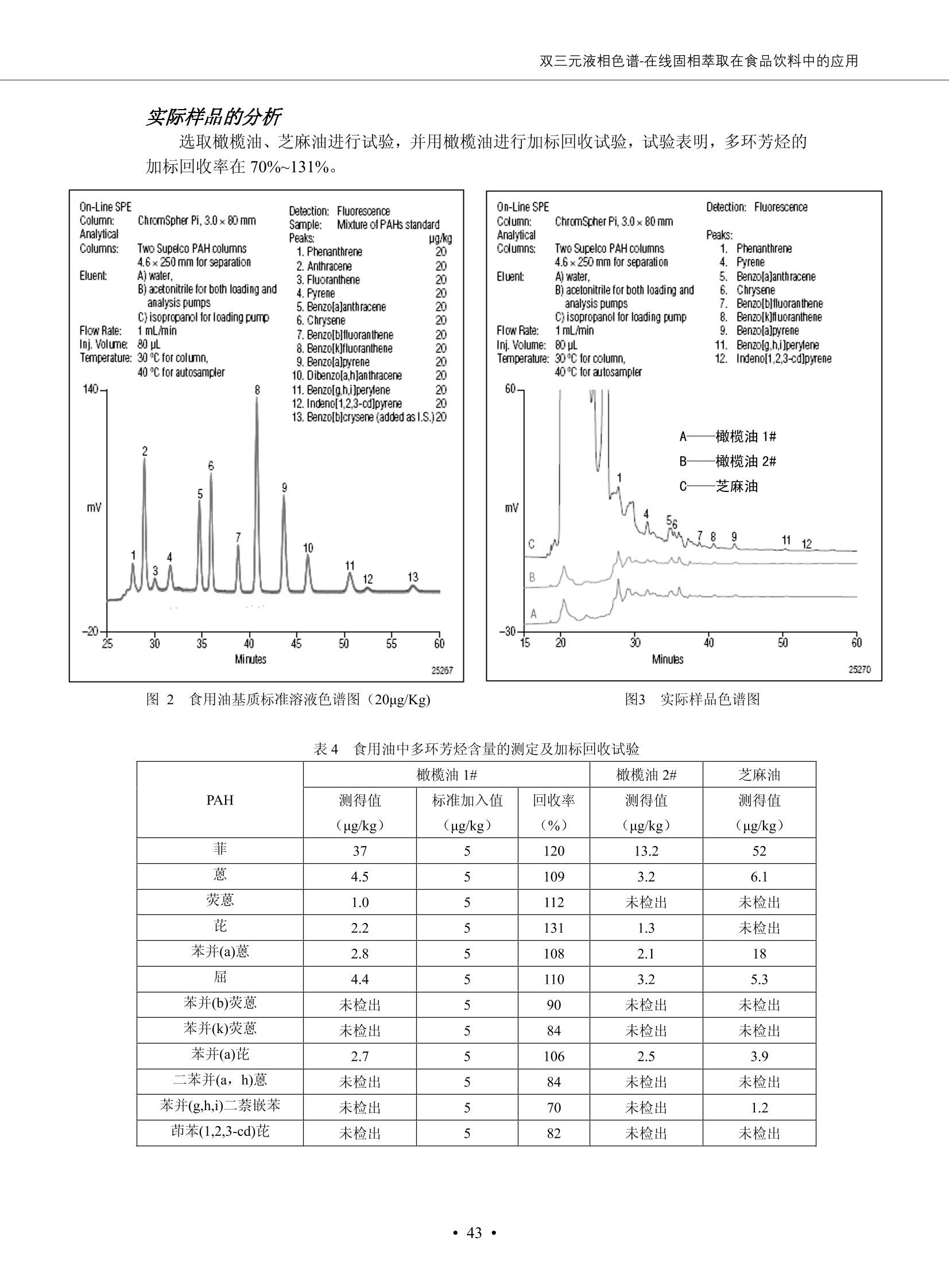
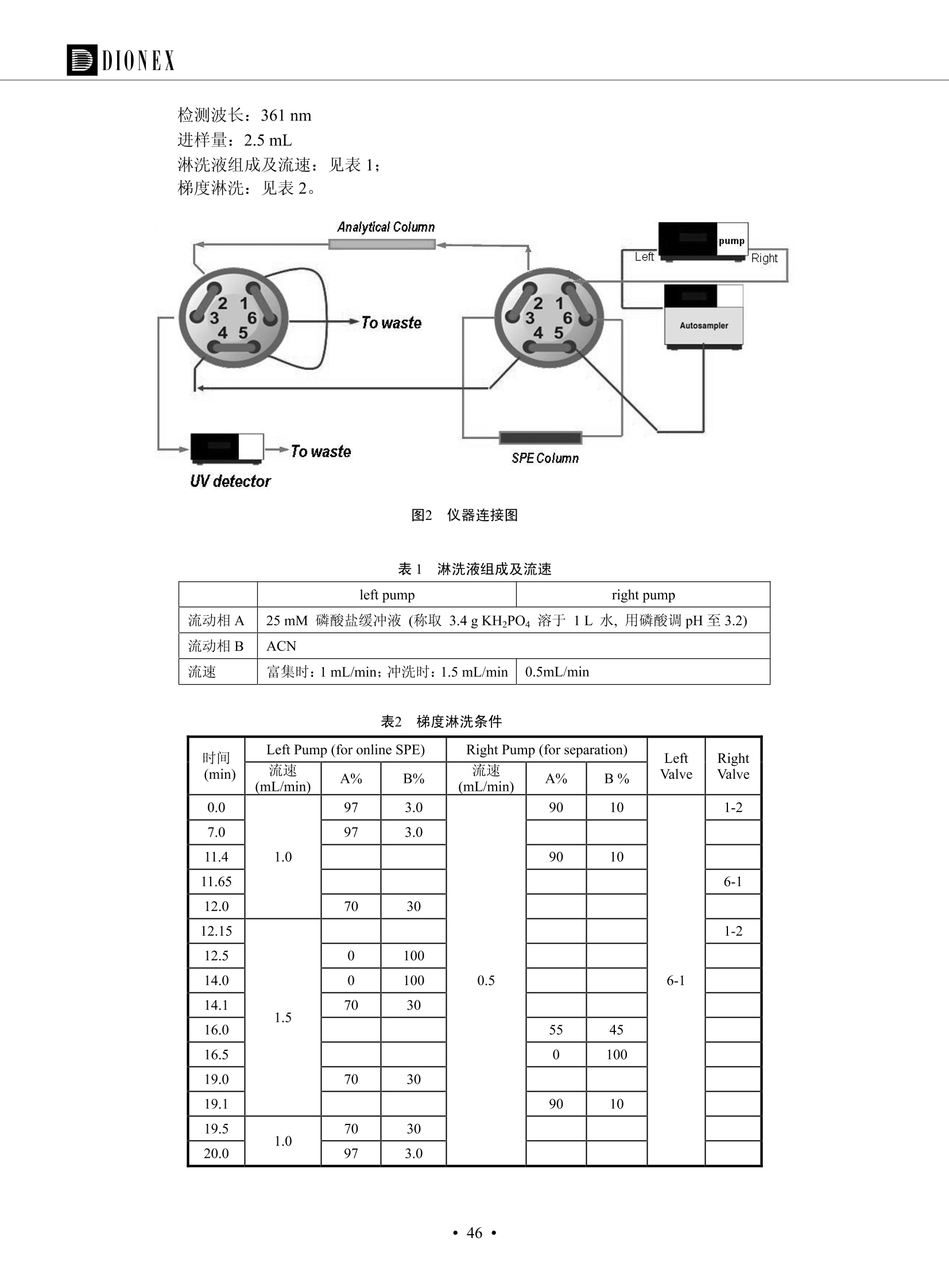
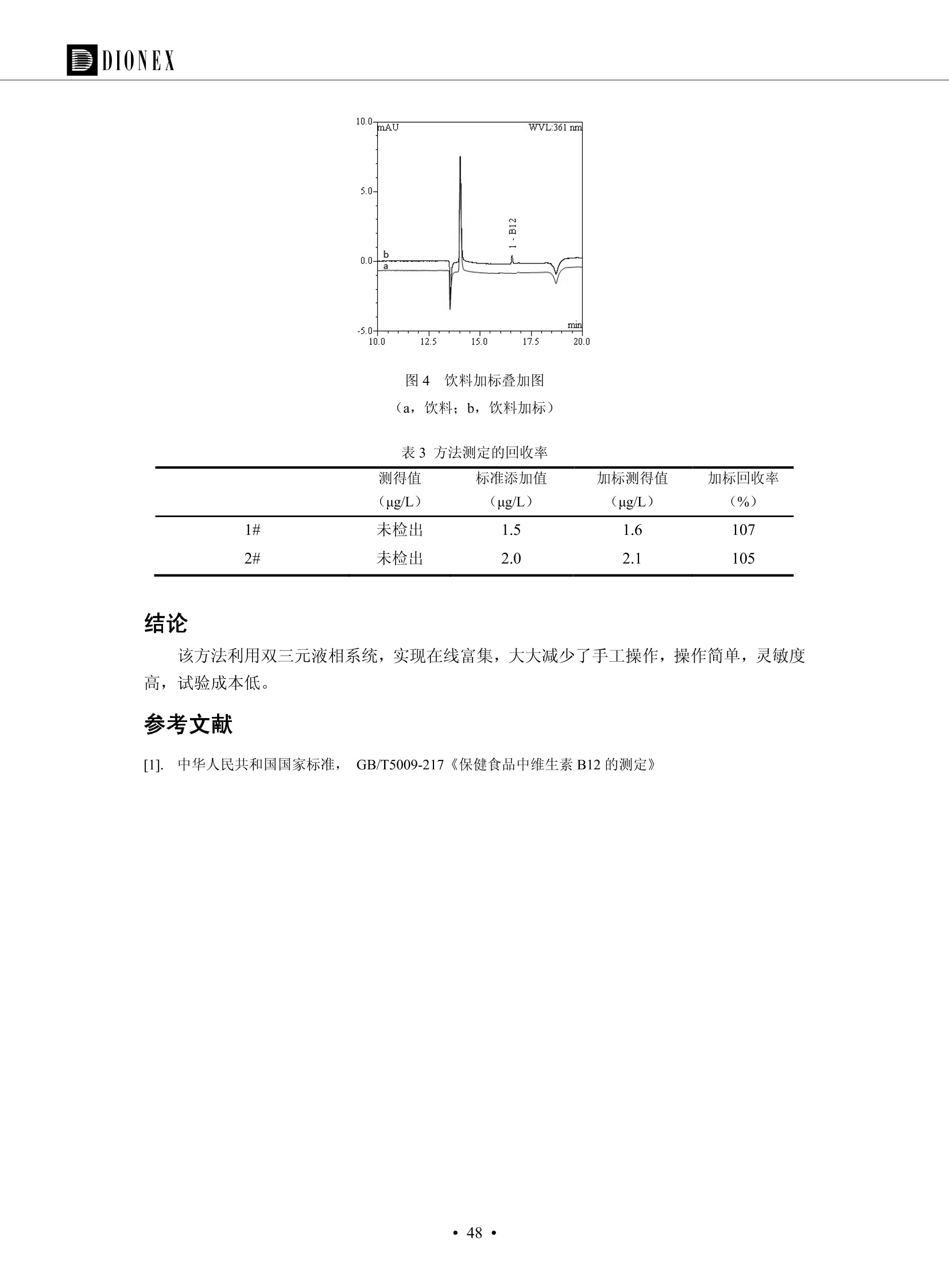
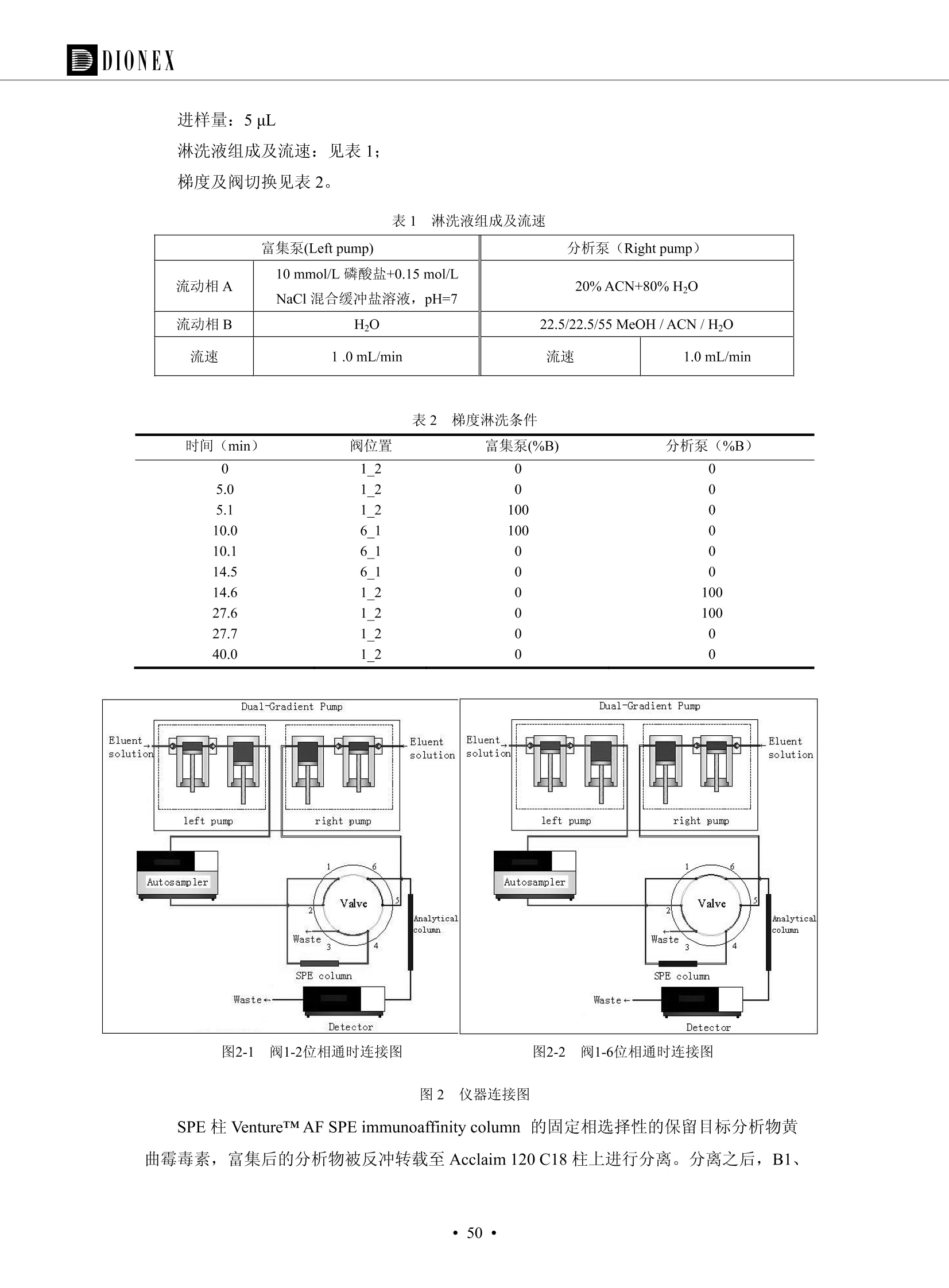
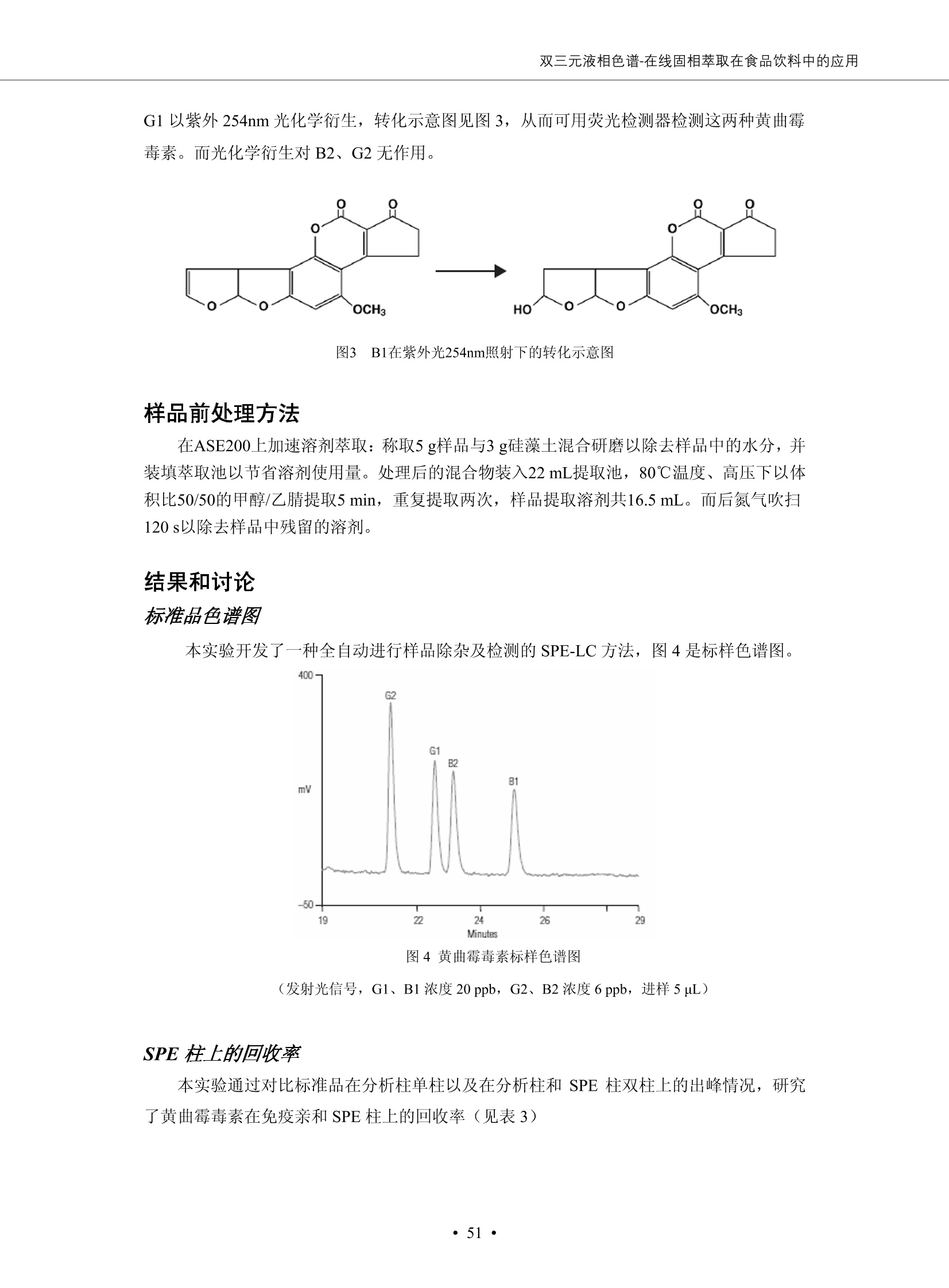
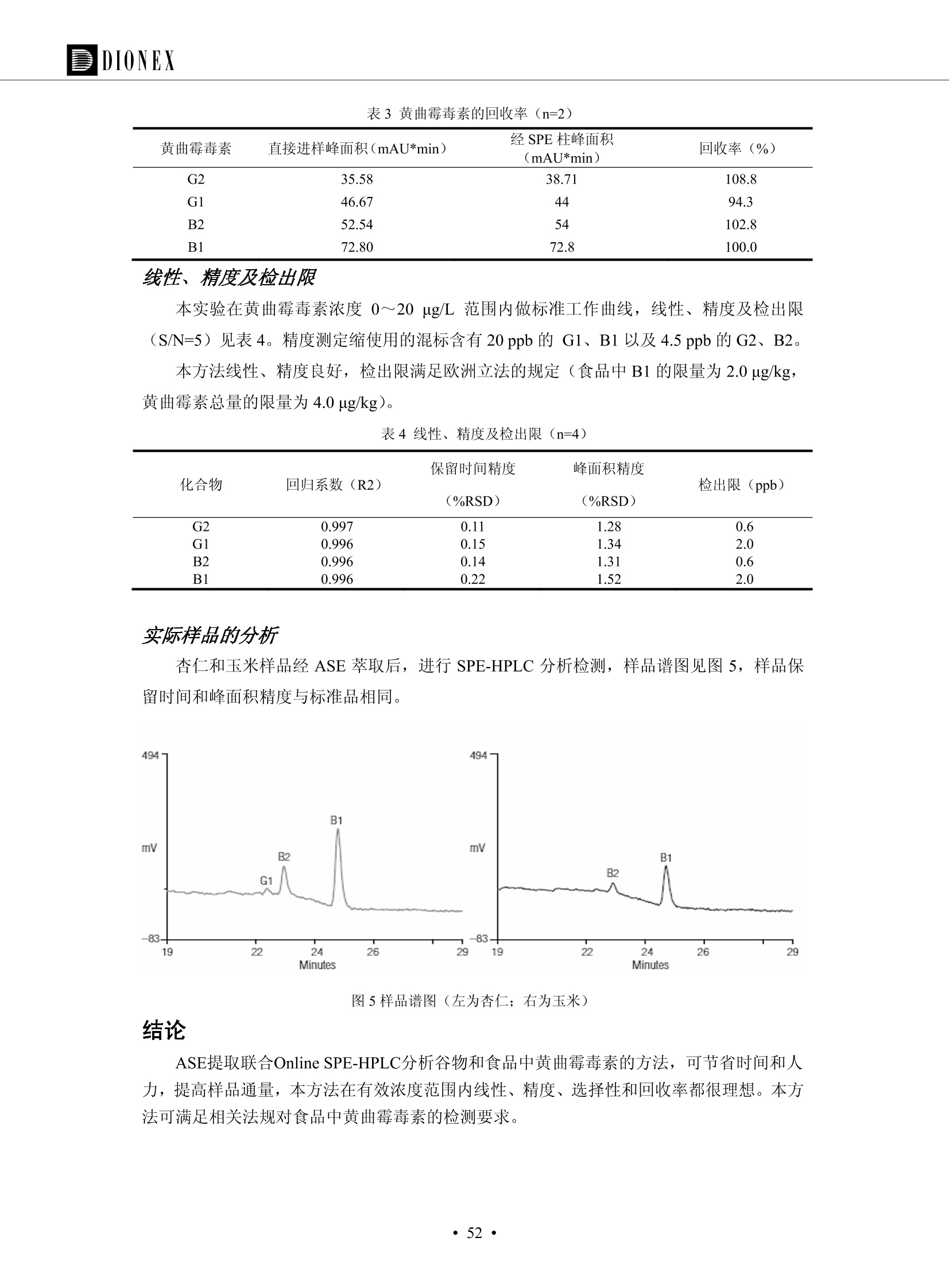
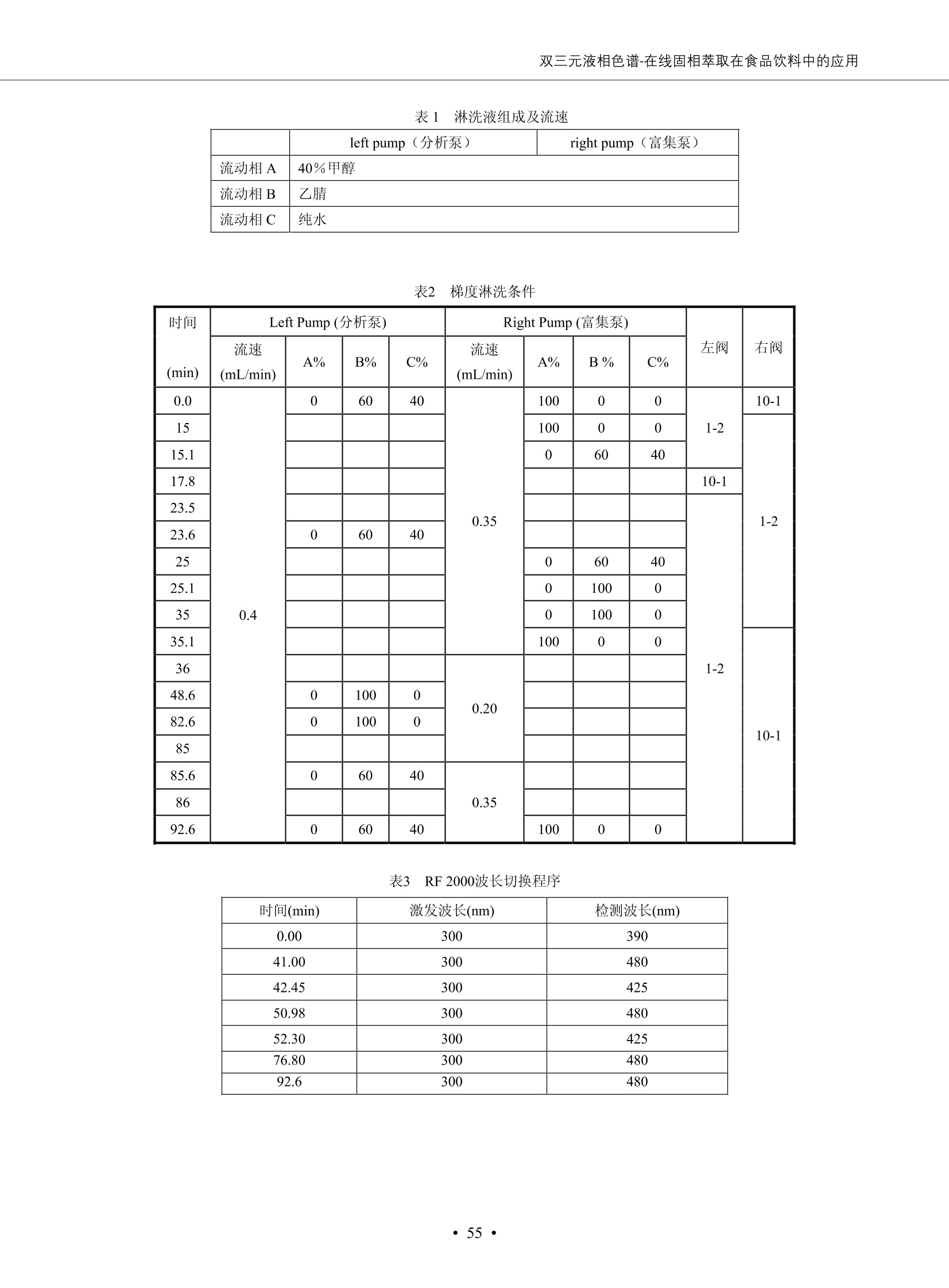
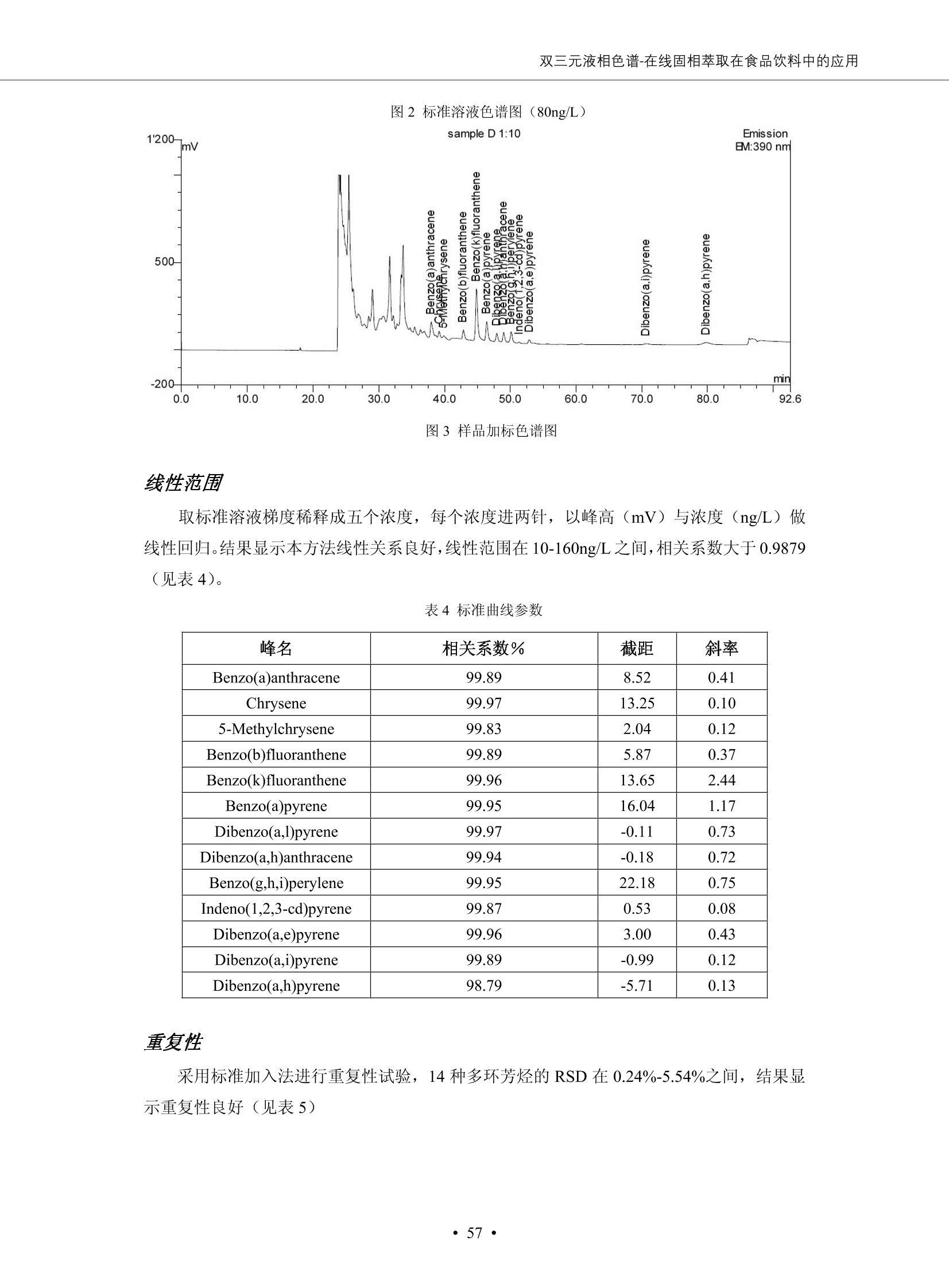
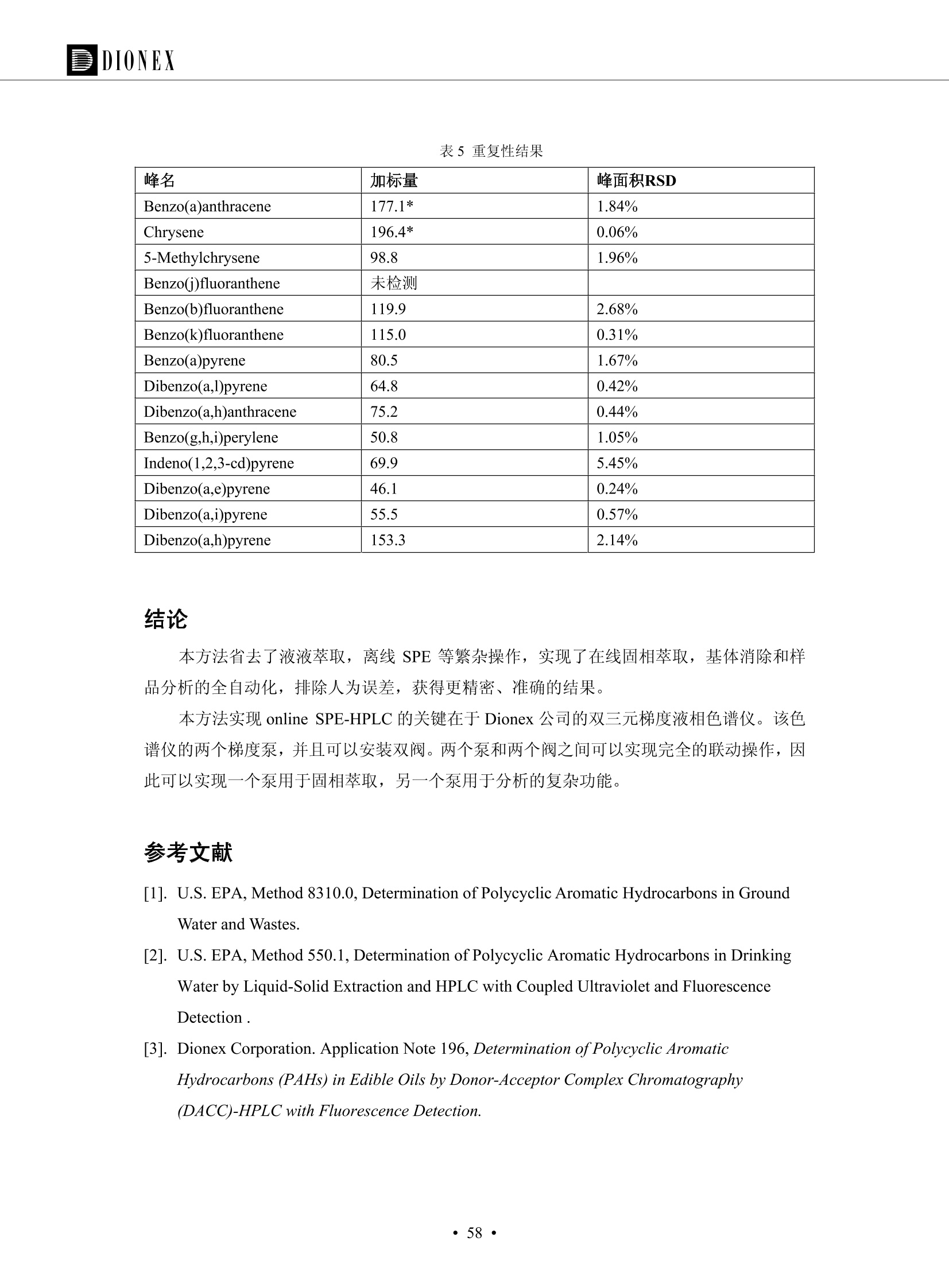
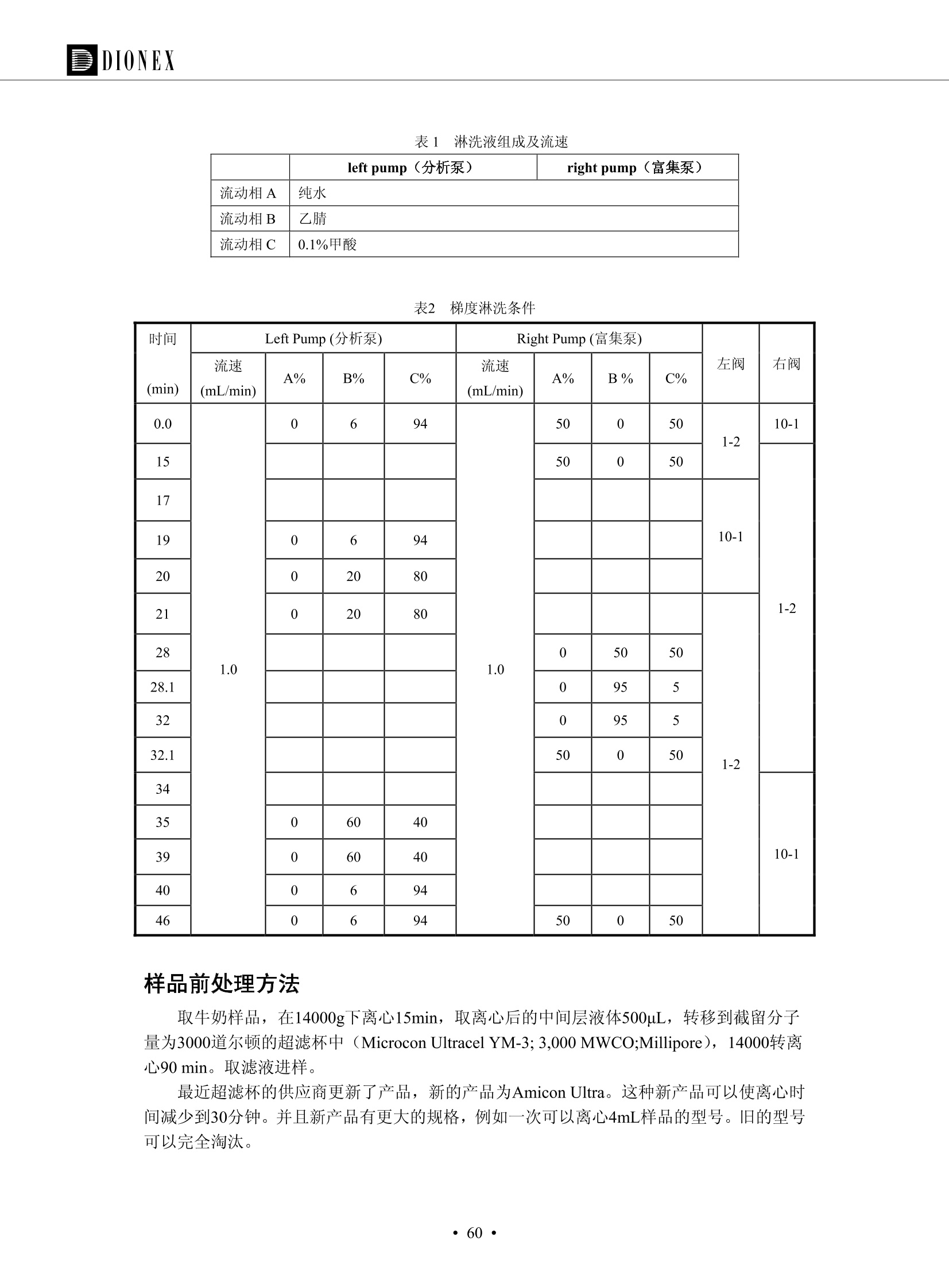
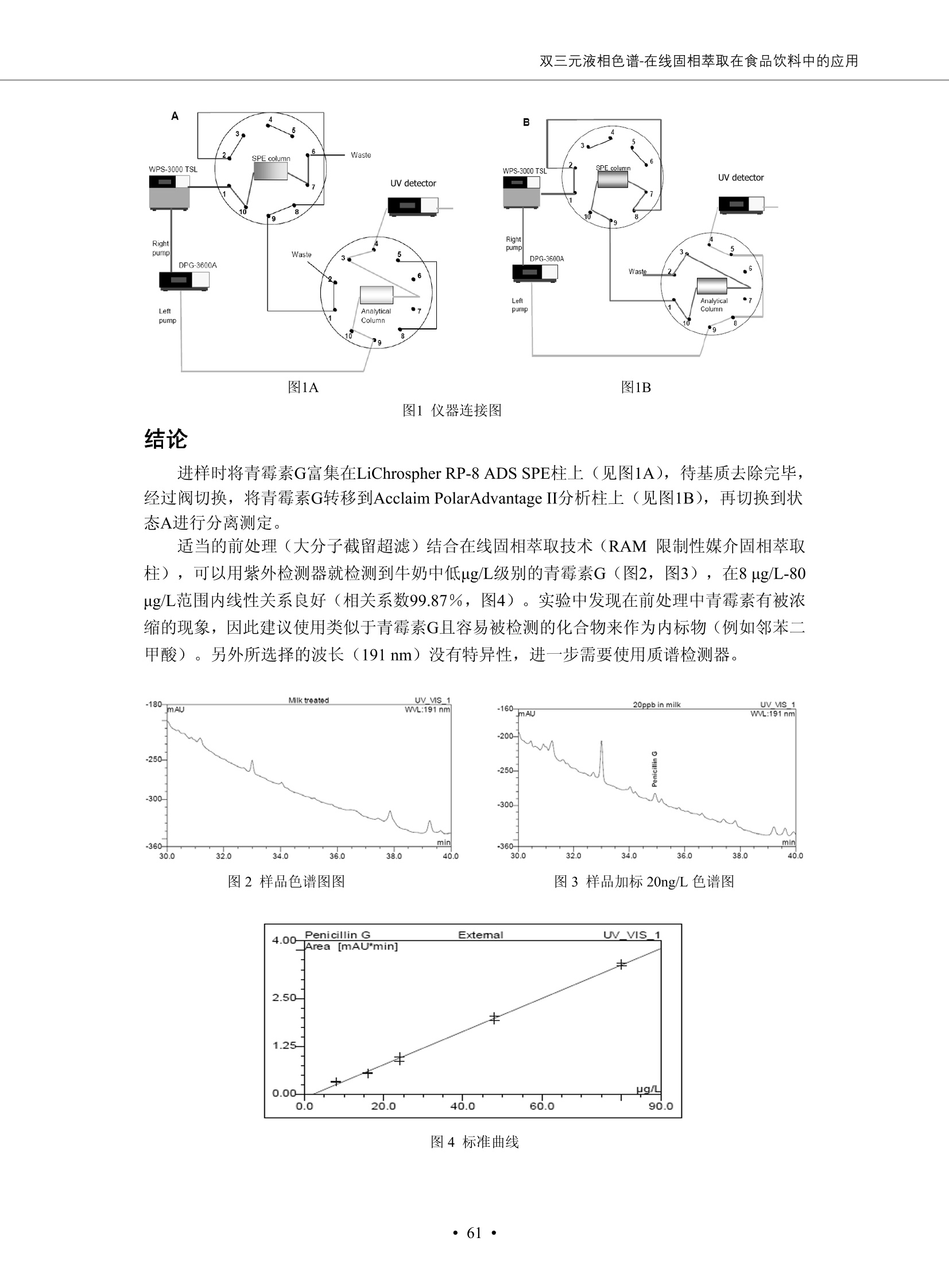
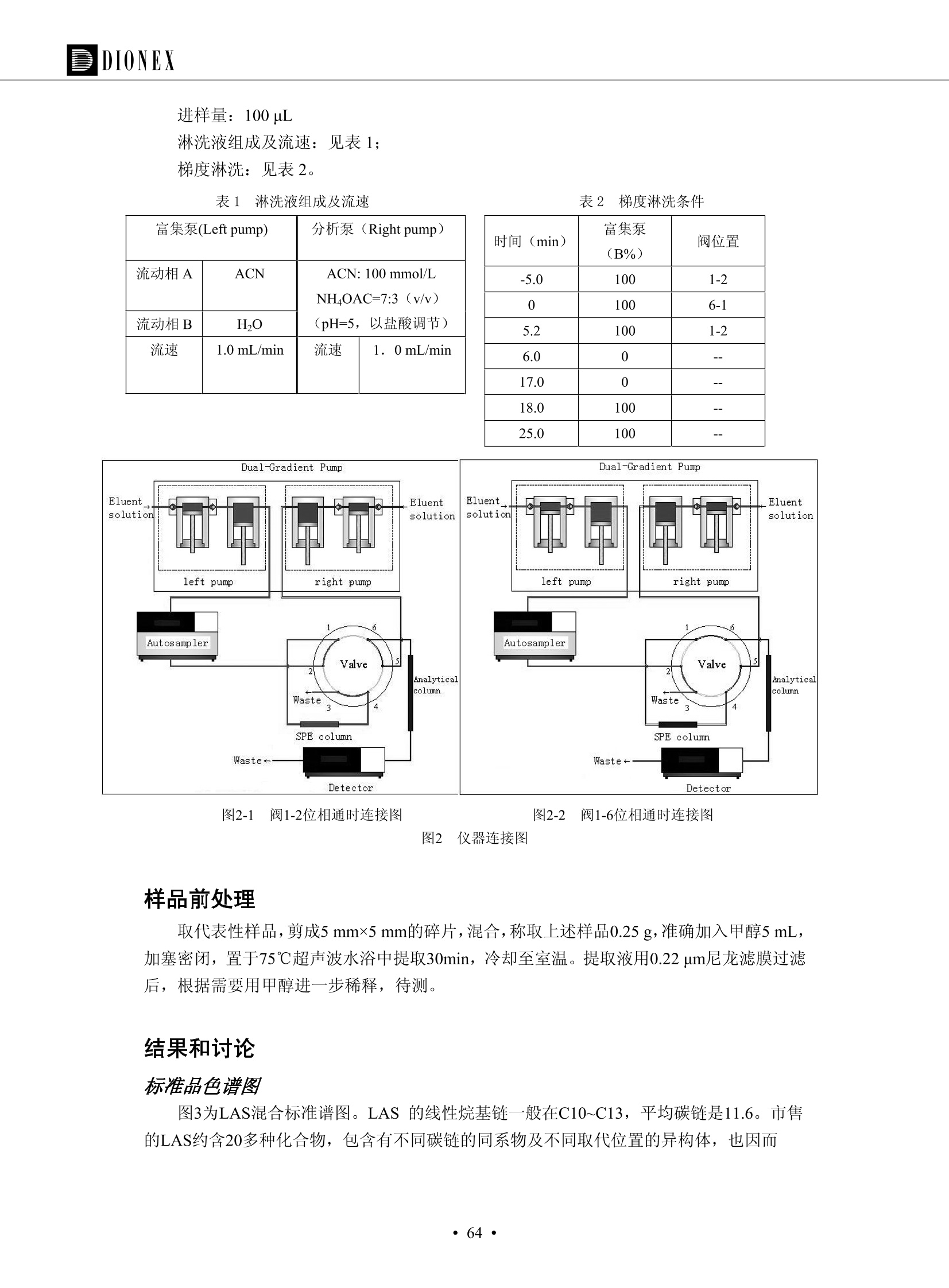
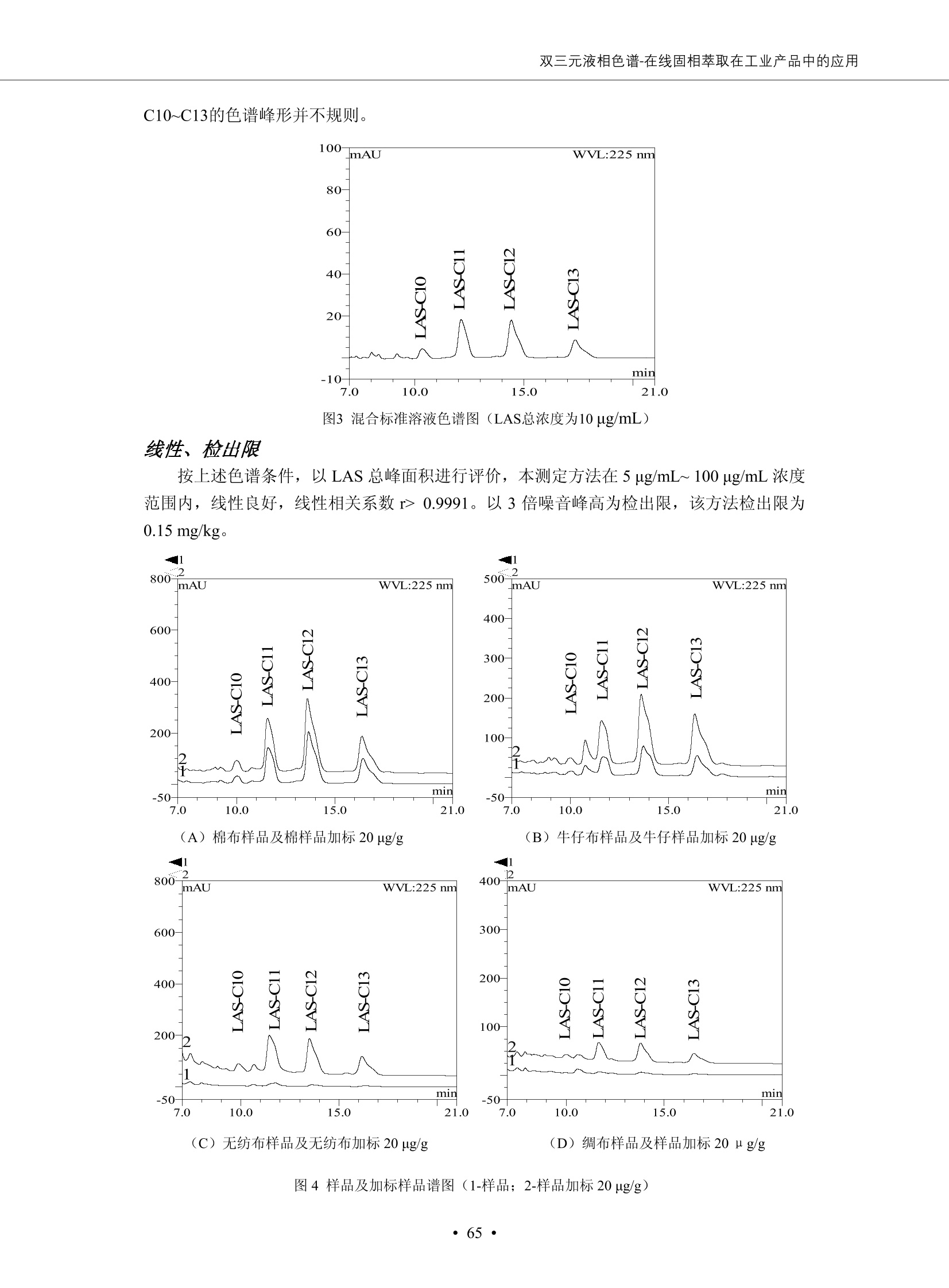
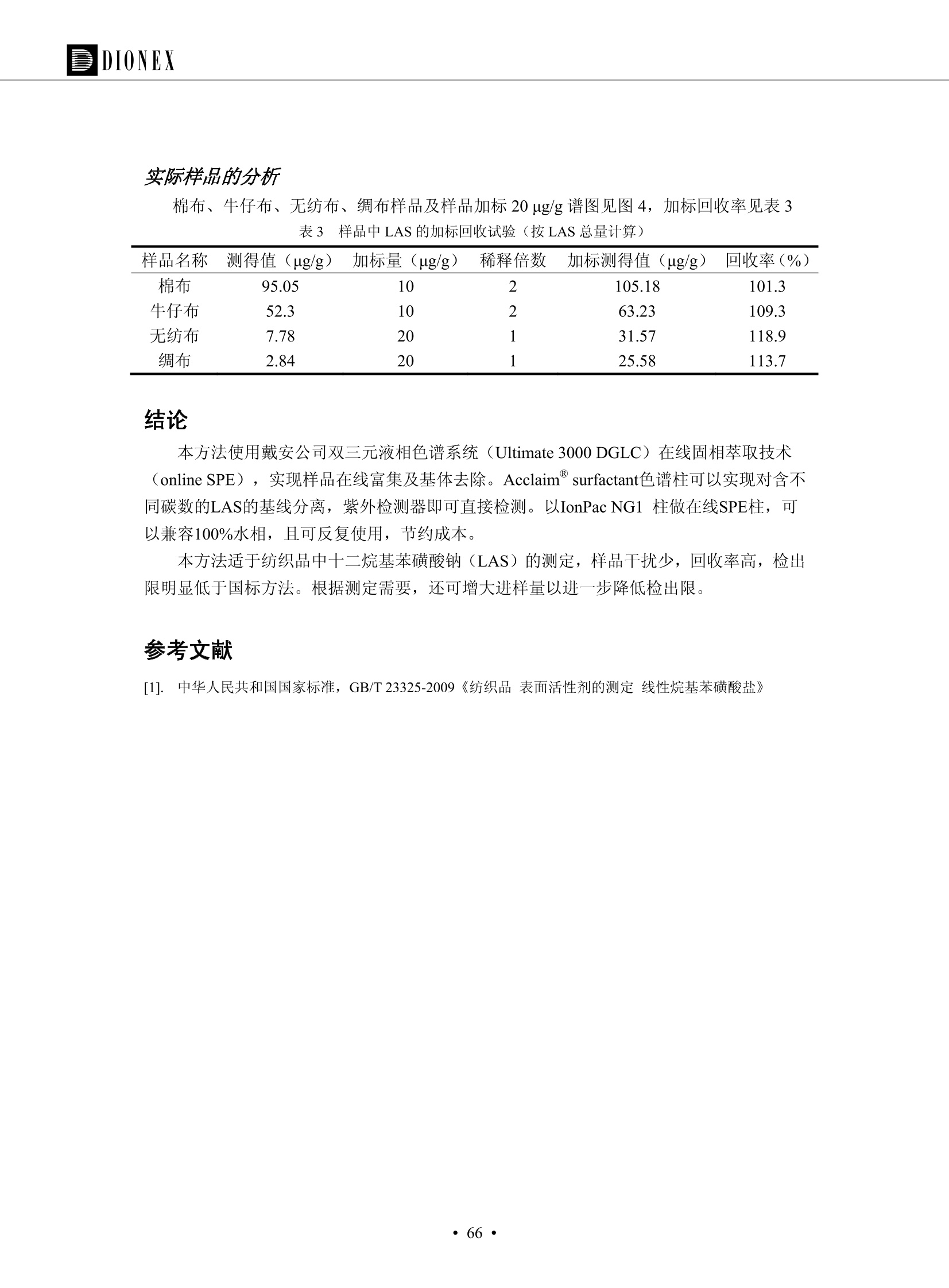
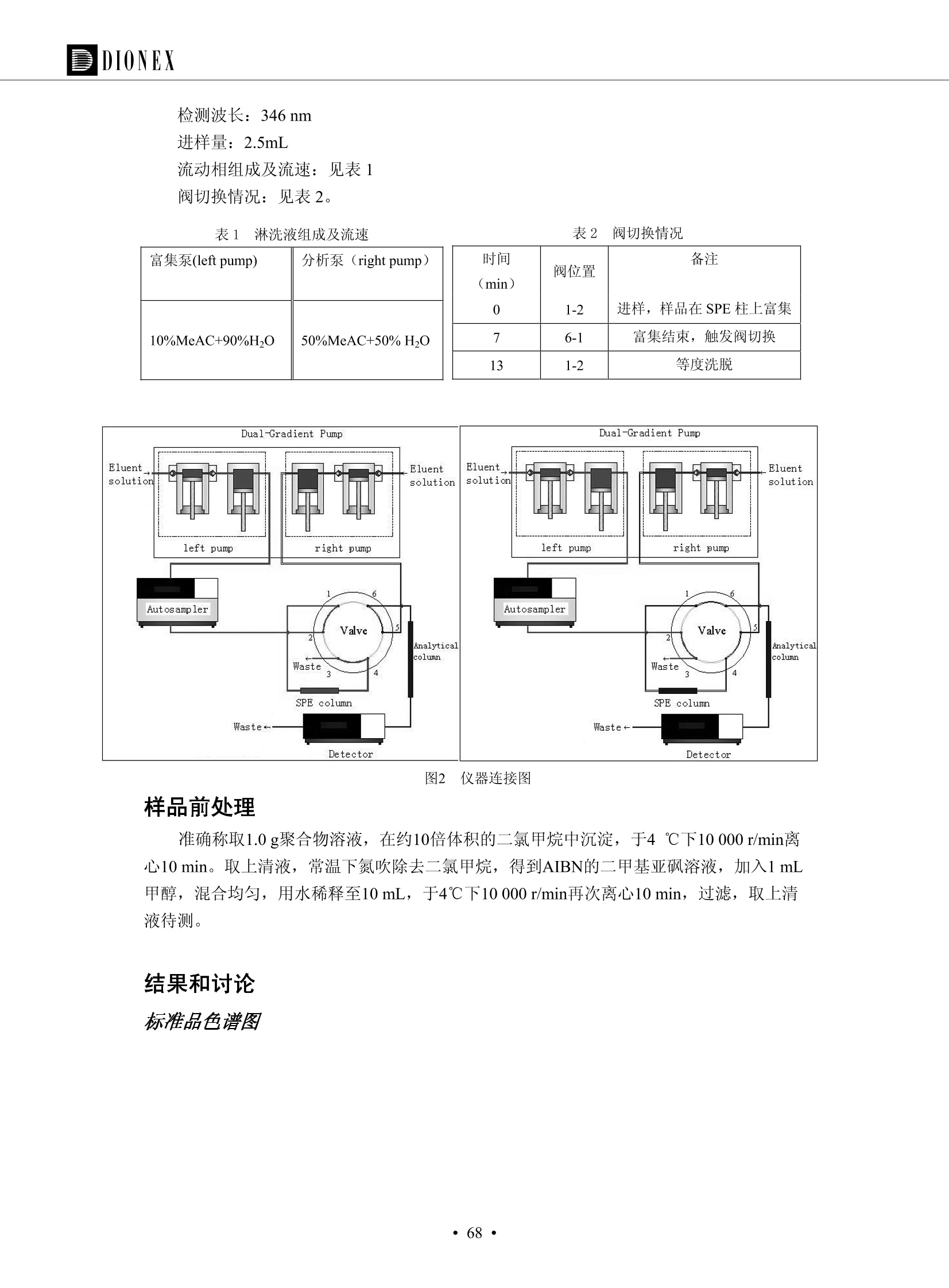
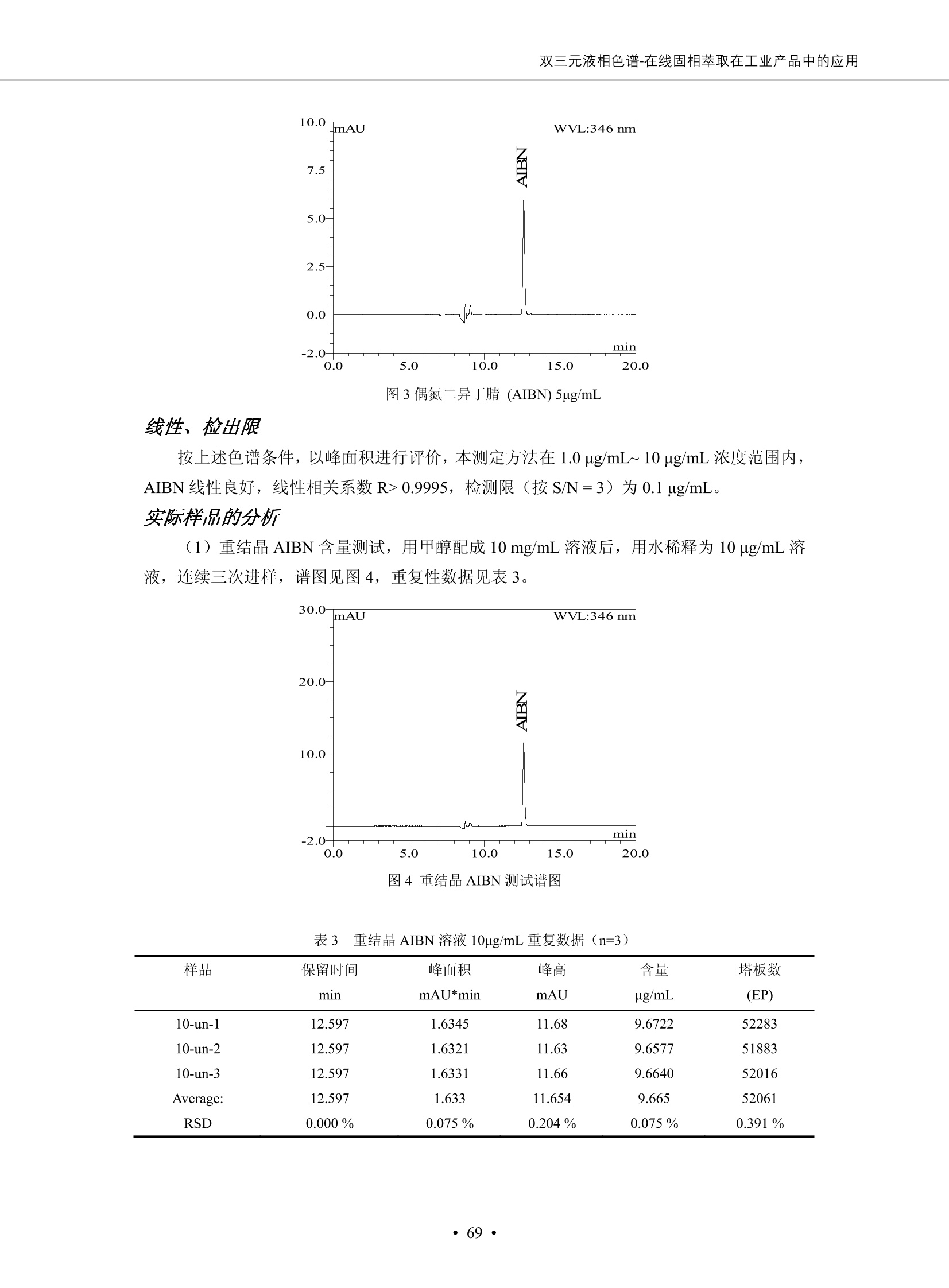
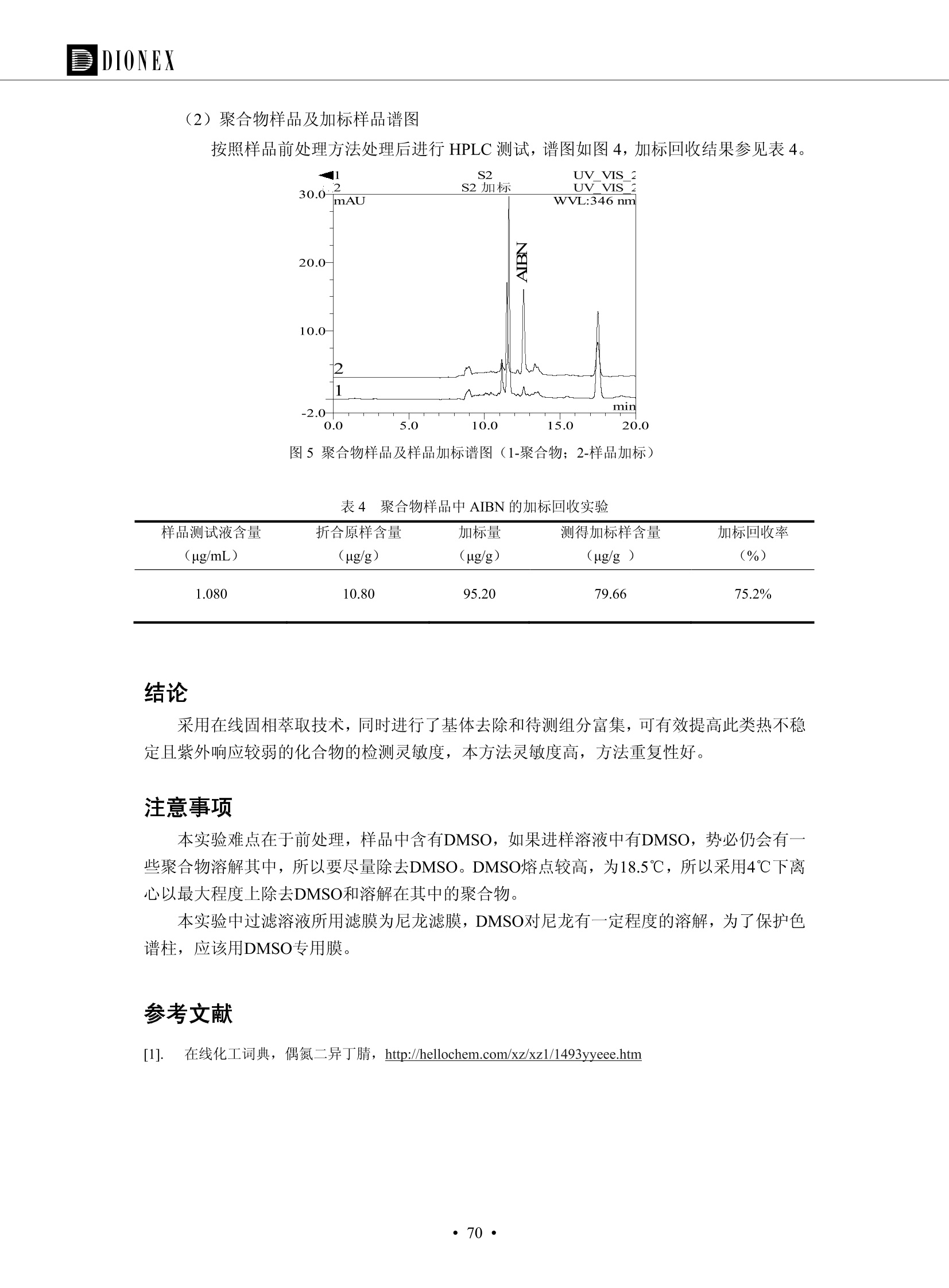
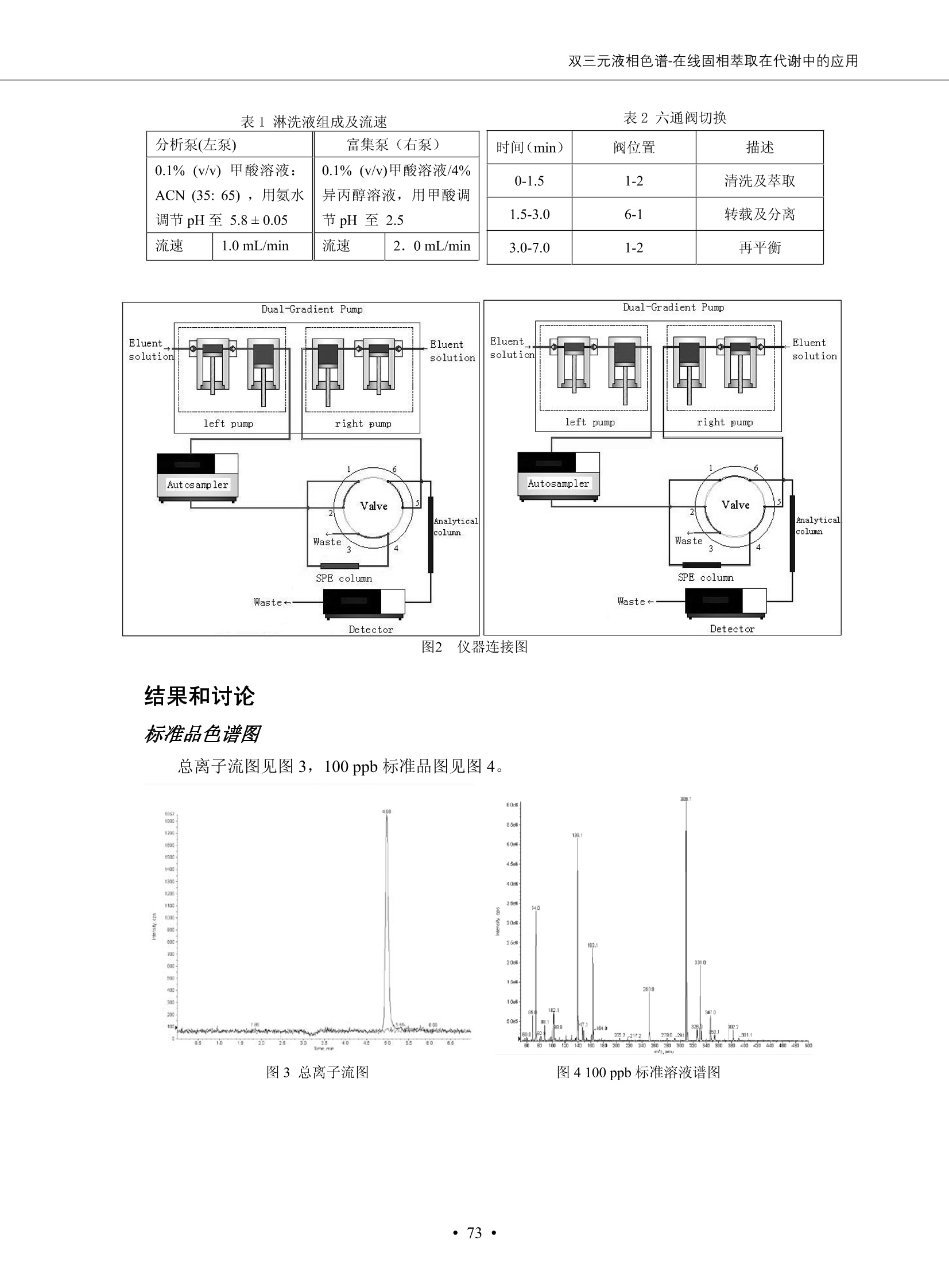
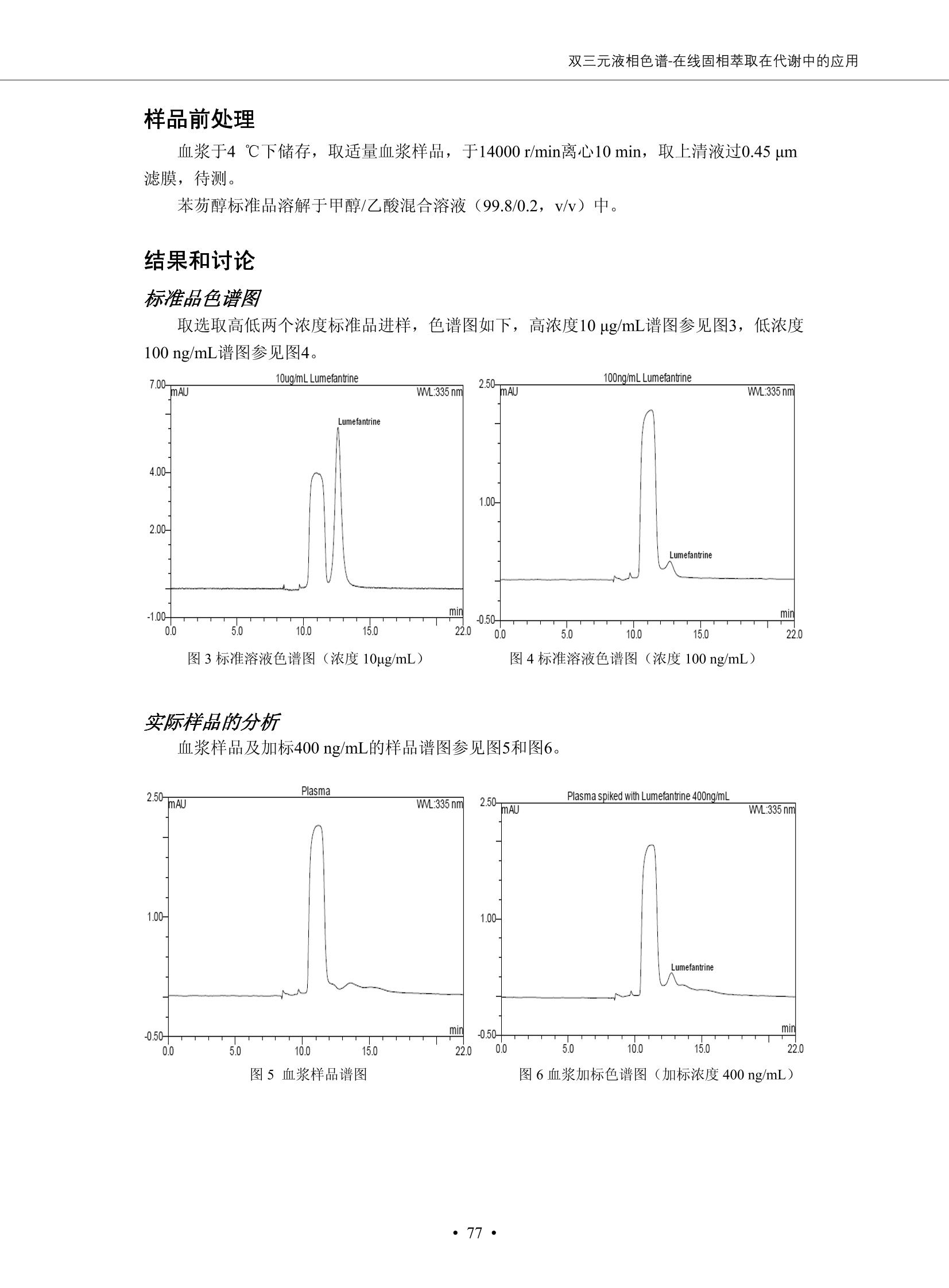
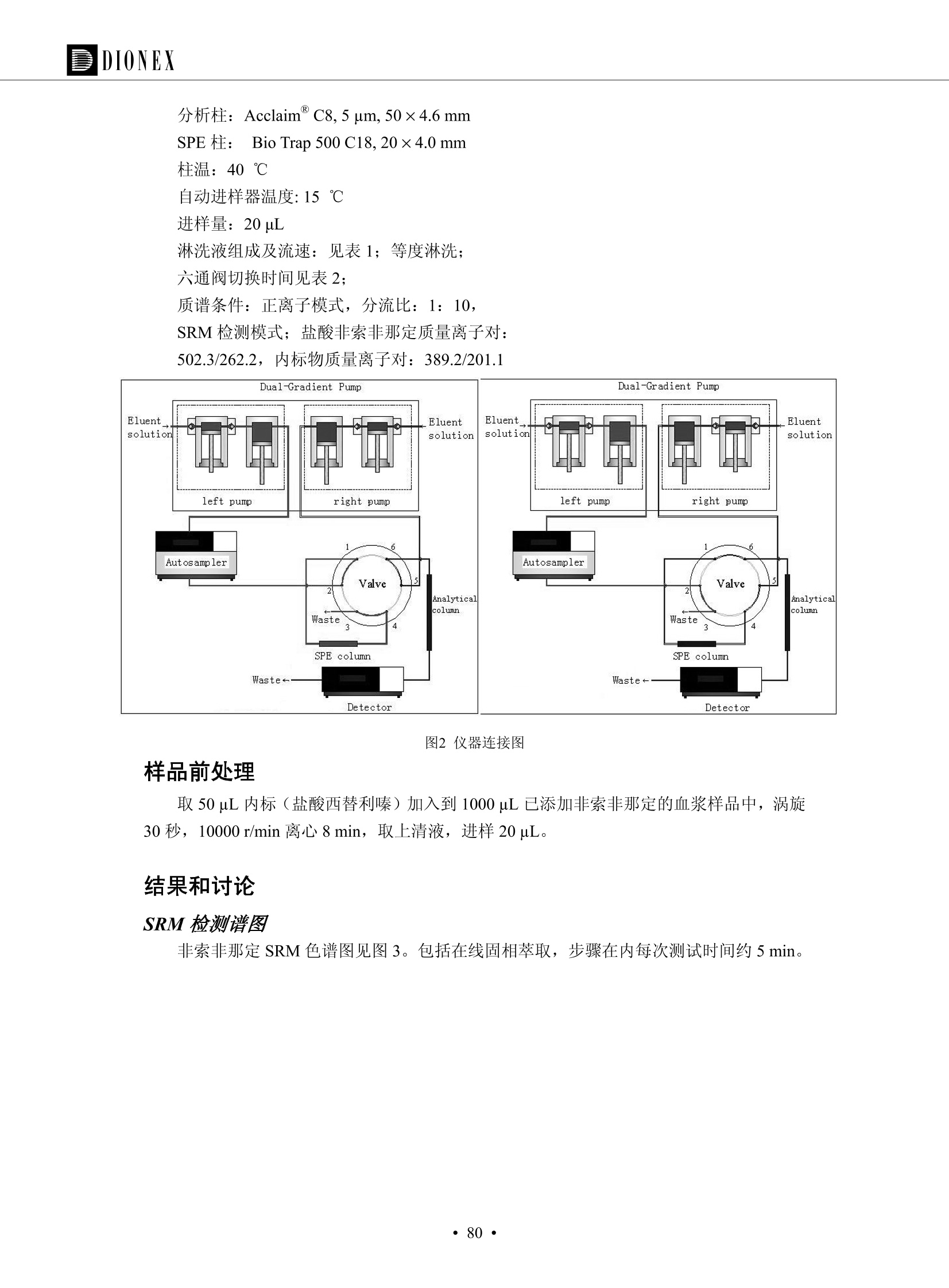
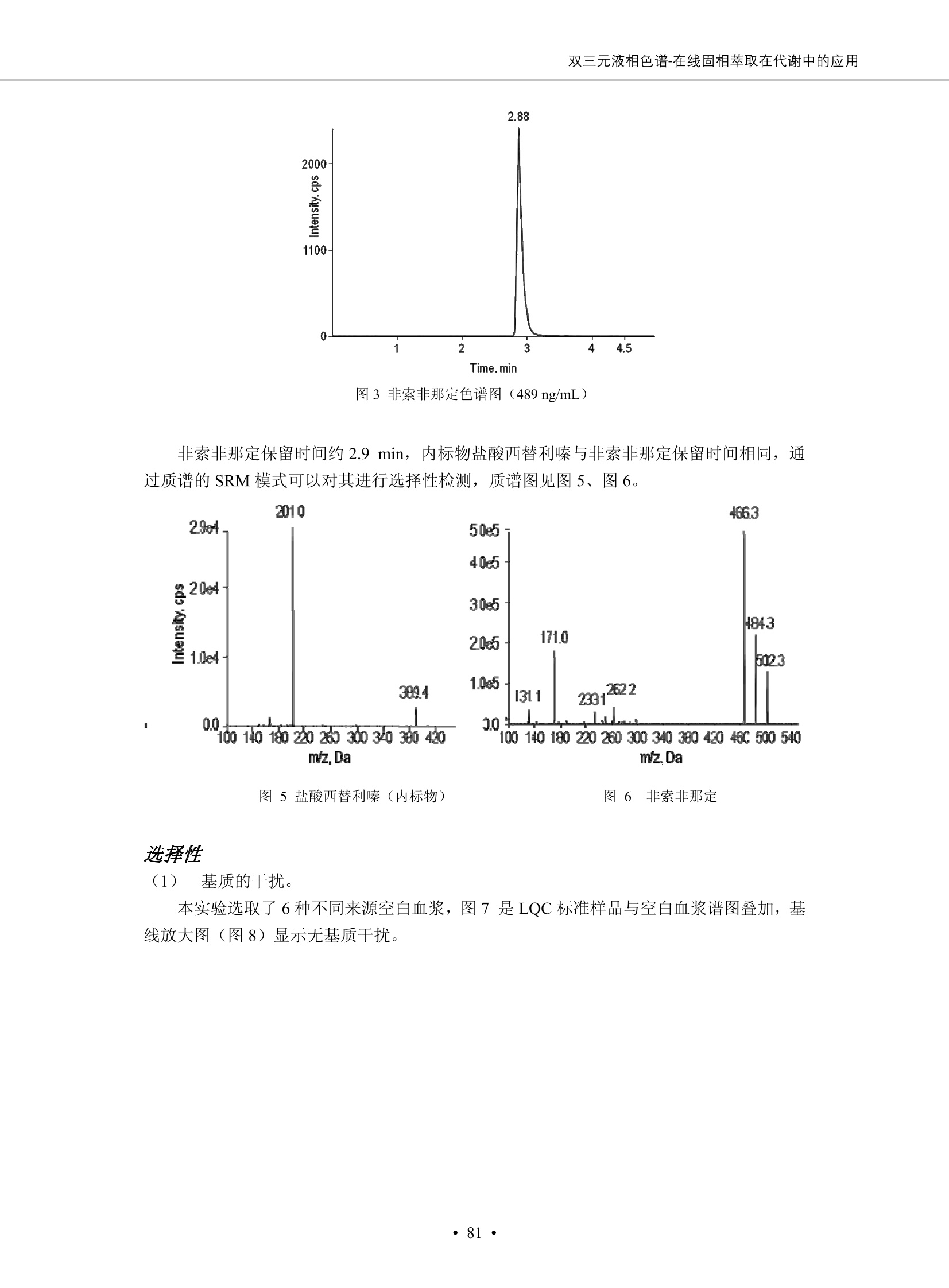
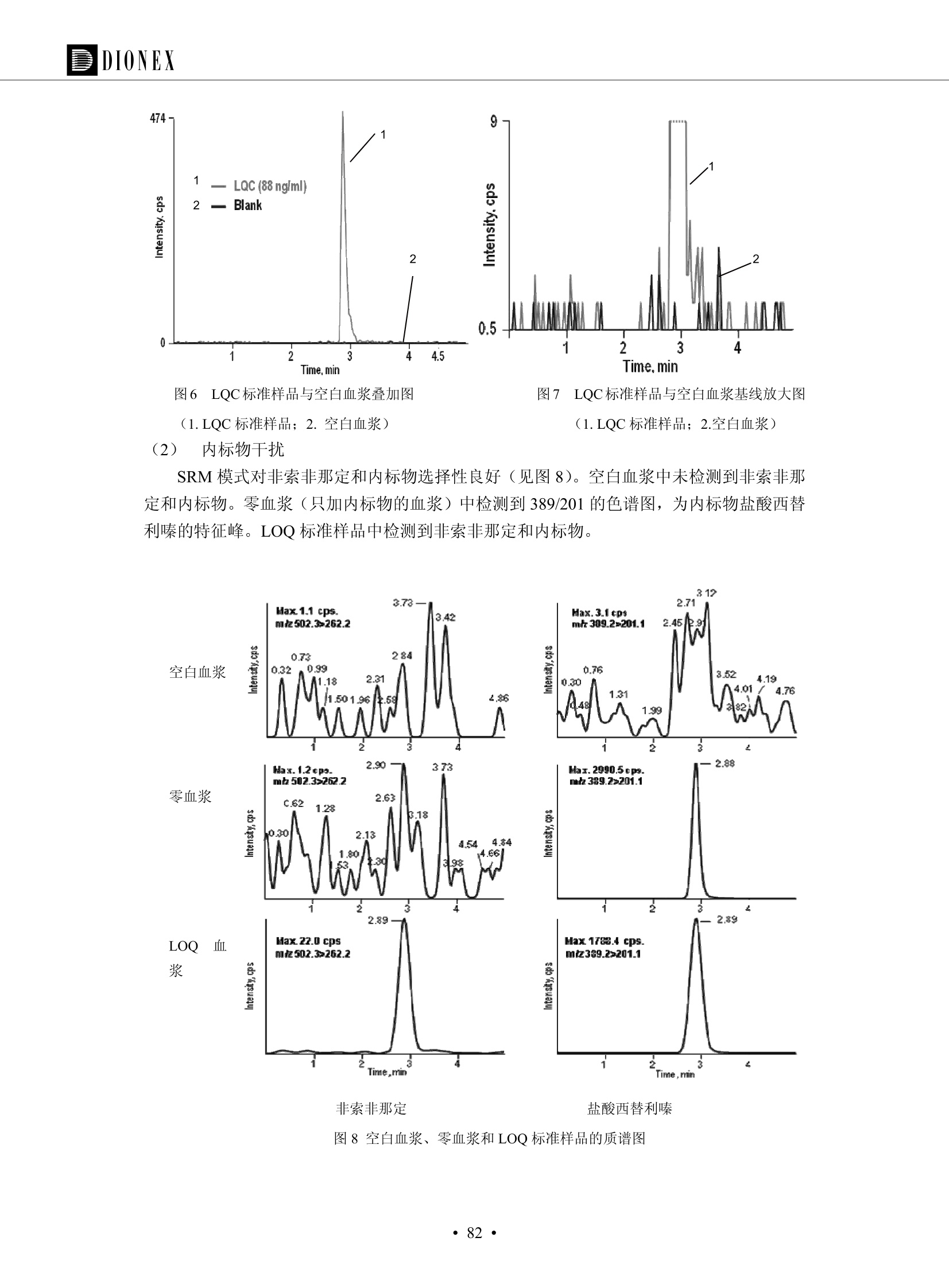
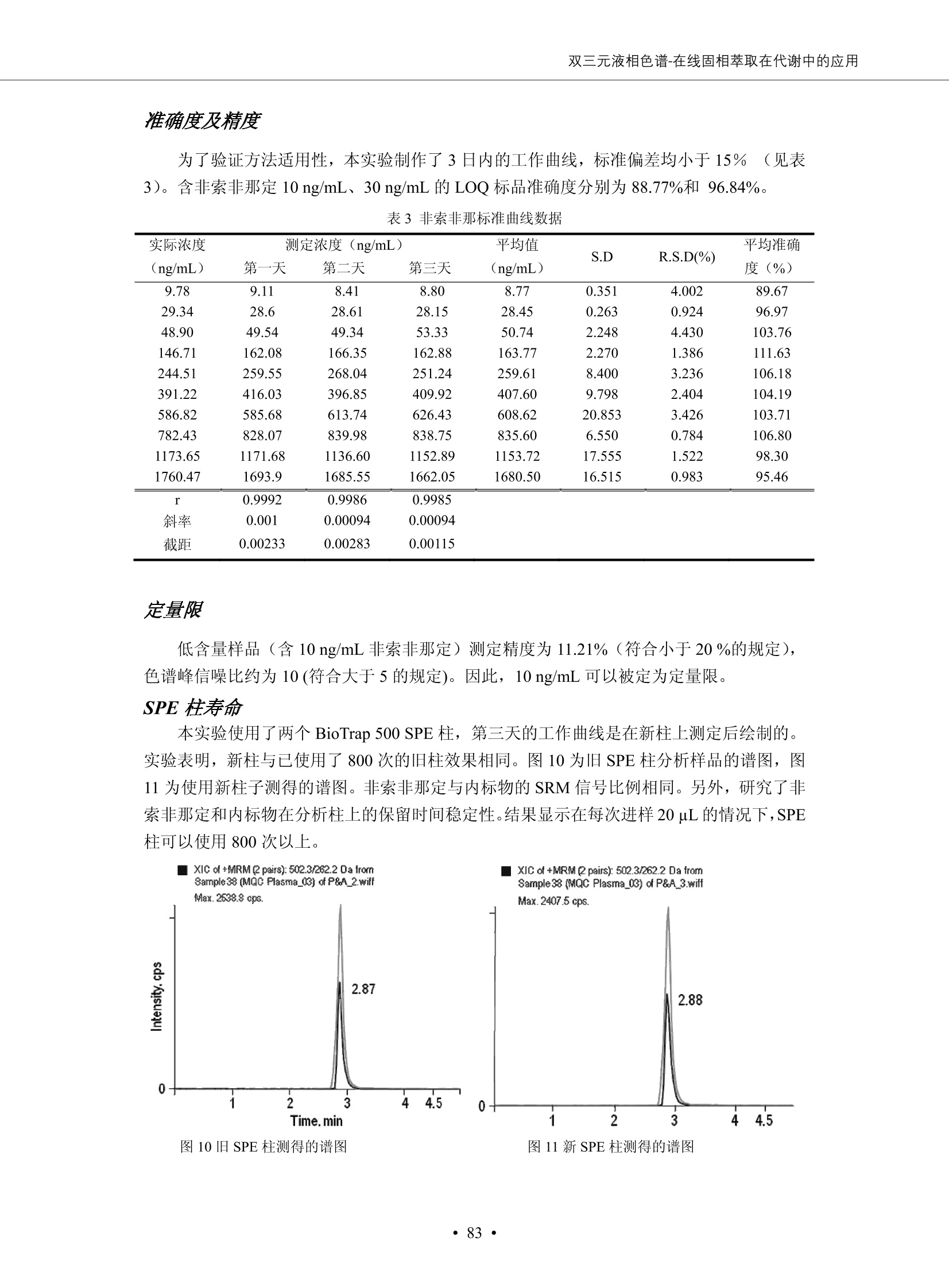
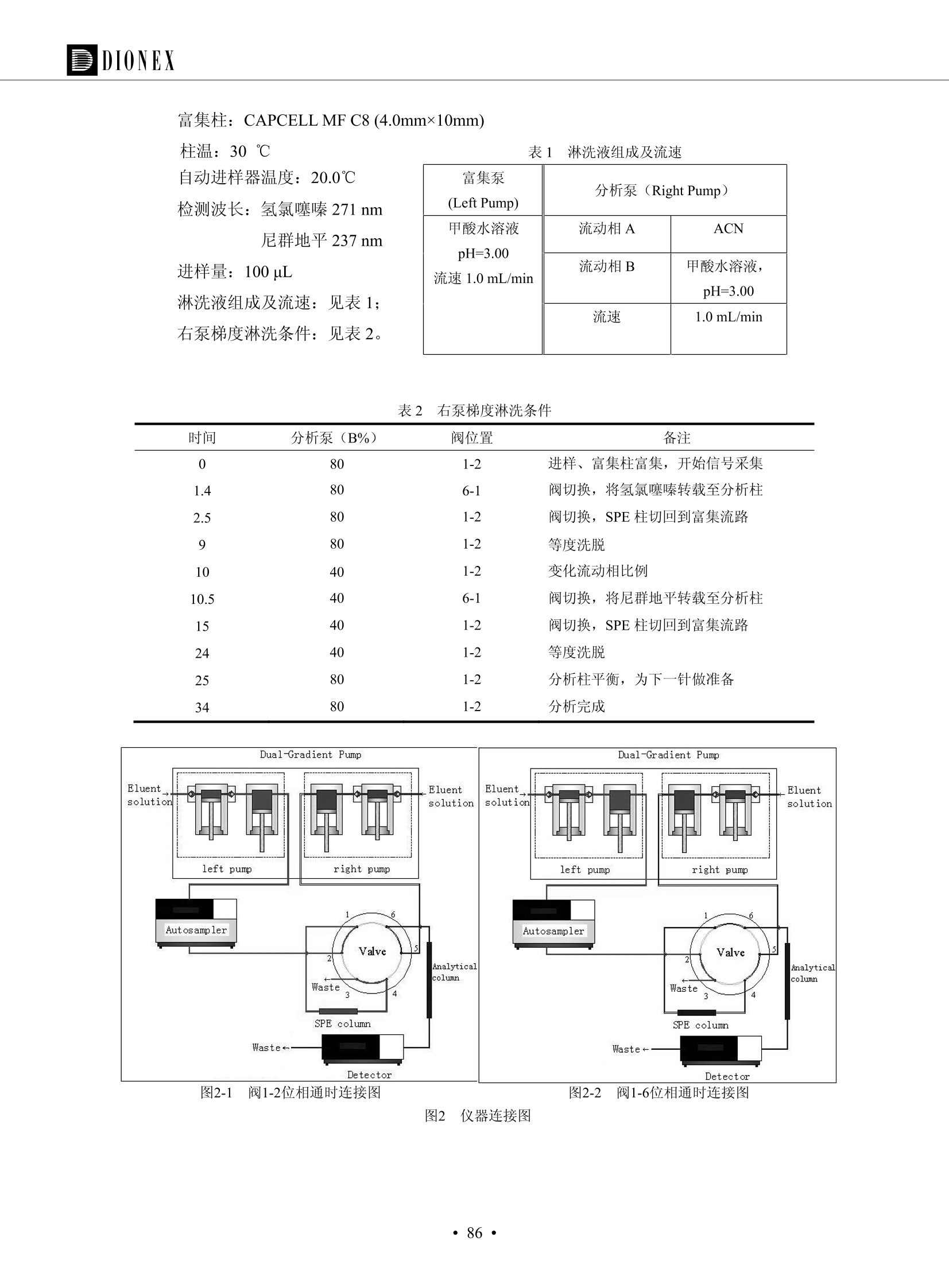
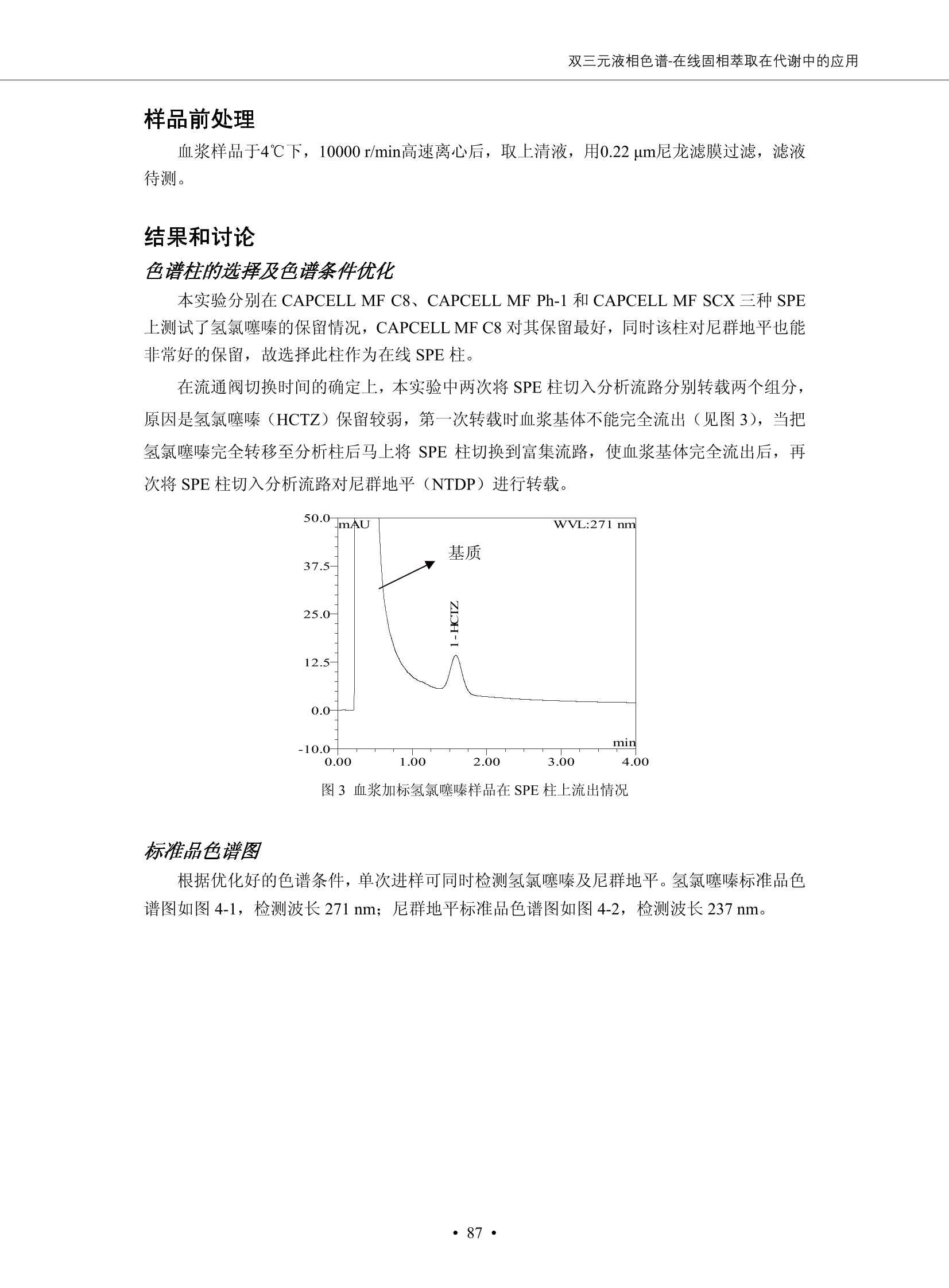
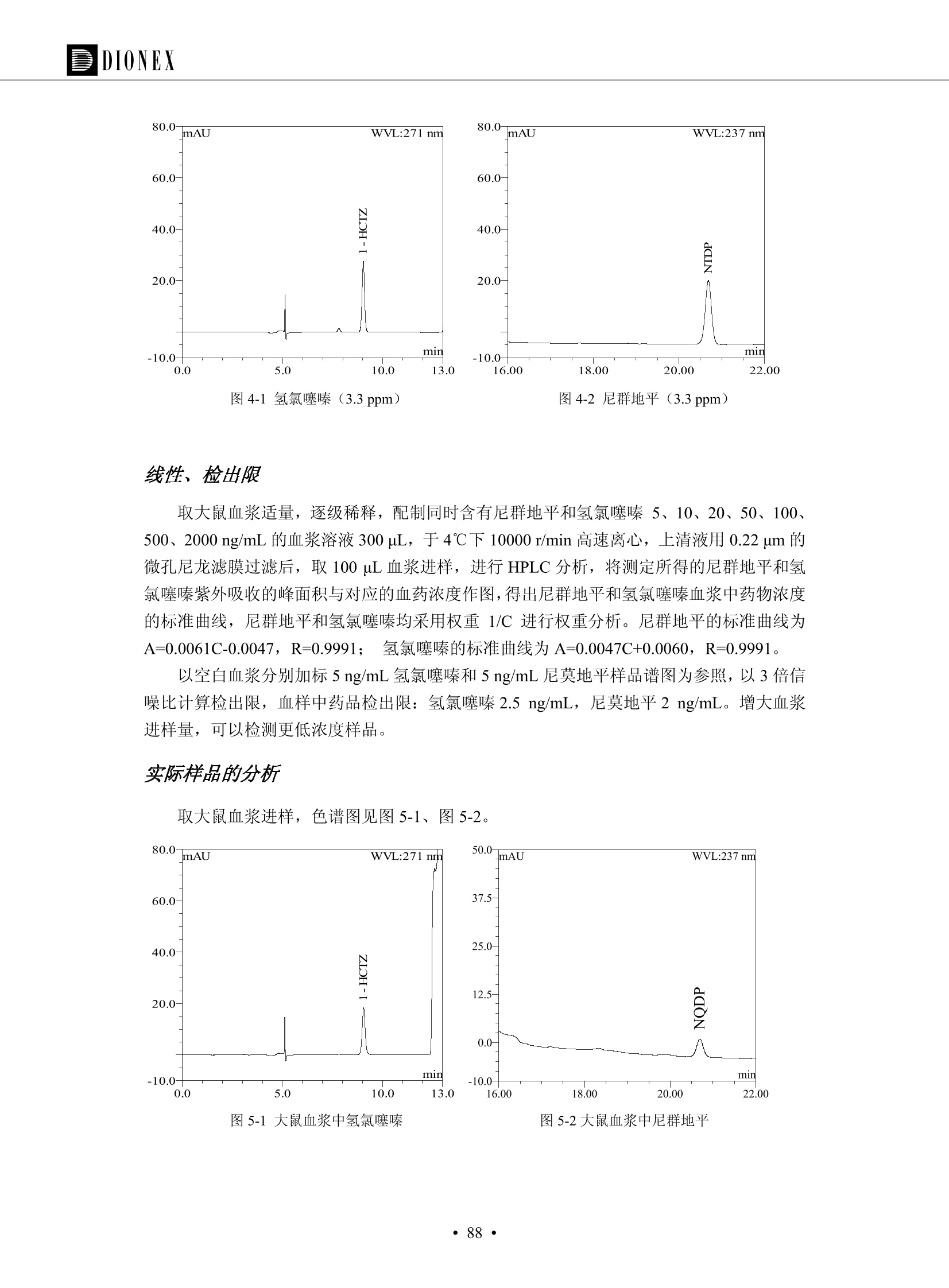
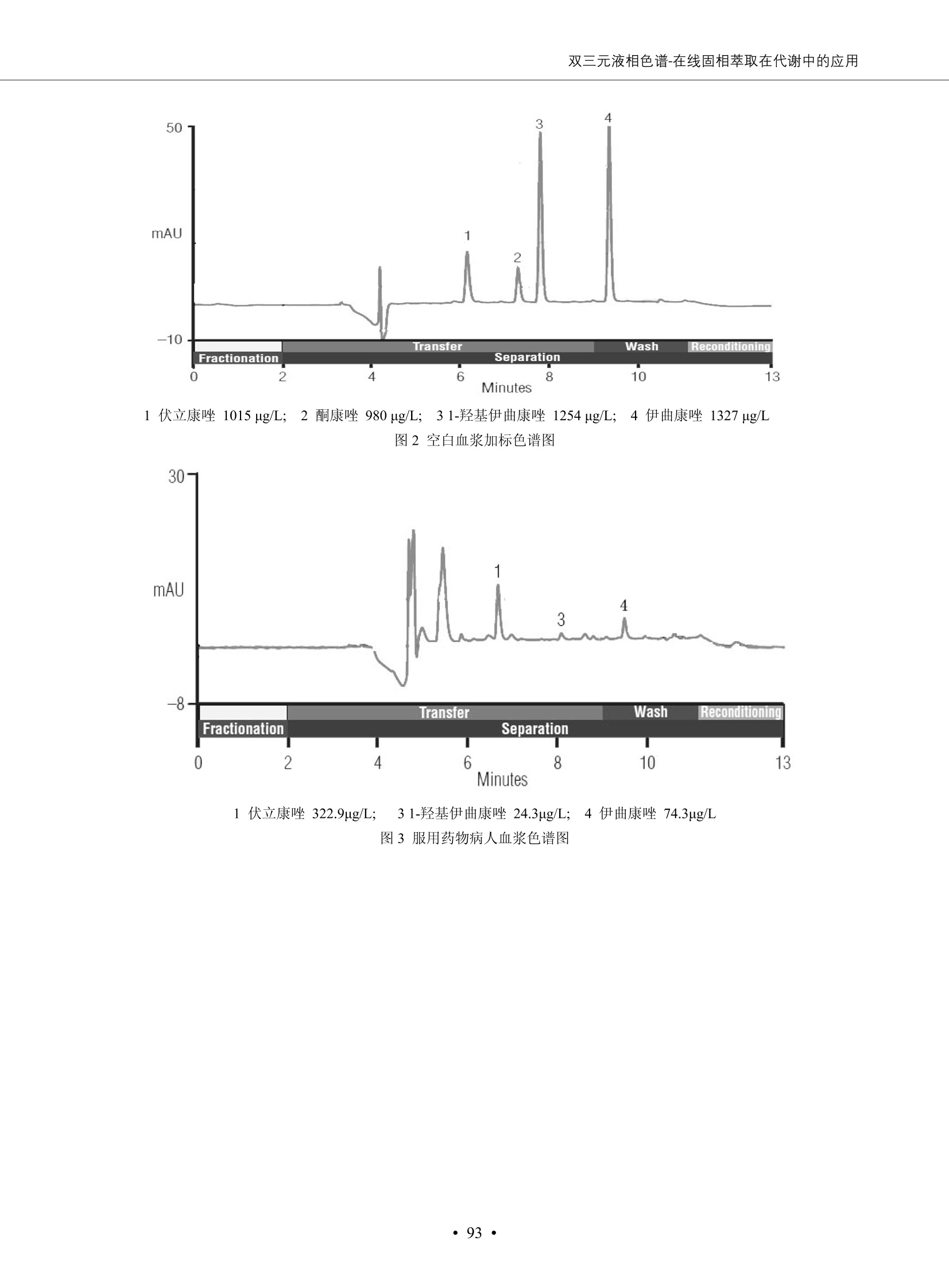
DIONEX 双三元液相色谱-在线固相萃取在环境领域的应用 戴安公司双三元液相应用文集(DGLC-AC) 环境、食品饮料、工业产品及药物代谢 戴安中国有限公司 目录 第一部分双三元液相色谱-在线固相萃取在环境领域中的应用. 2 DGLC-E-A01在线固相萃取-高效液相色谱-紫外和荧光检测联用测定自来水中的多环芳烃....... 3 DGLC-E-A02 在线固相萃取-高效液相色谱-紫外检测法测定饮用水和瓶装矿水水中苯酚... 8 DGLC-E-A03 在线固相萃取-高效液相色谱-紫外检测法测定水样中痕量微囊藻毒素 .... 16 DGLC-E-A04 在线固相萃取-高效液相色谱-紫外检测法测定环境水样中四种痕量邻苯二甲酸酯. 21 DGLC-E-A05在线固相萃取-高效液相色谱-荧光检测法测定污水处理厂水样中直链烷基苯磺酸盐 (LAS) ....26 DGLC-E-A06 在线固相萃取-高效液相色谱-紫外检测法测定污水处理厂水样中直链烷基苯磺酸盐 (LAS)....31DGLC-E-A07在线固相萃取-高效液相色谱-紫外检测法测定水样中痕量莠去津(阿特拉津) 35 第二部分双三元液相色谱-在线固相萃取在食品饮料中的应用. ....39 DGLC-F-A01在线固相萃取-高效液相色谱-荧光检测法测定食用油中多环芳烃3. .40 DGLC-F-A02 在线固相萃取-高效液相色谱-荧光检测法测定饮料中维生素B12.... .45 DGLC-F-A03 加速溶剂萃取-在线固相萃取-高效液相色谱-荧光检测法快速测定谷物或食品中的黄曲酶霉毒素...49 DGLC-F-A04 在线固相萃取-高效液相色谱-荧光检测法测定白兰地中的多环芳烃..... ....54 DGLC-F-A05 在线固相萃取-高效液相色谱-紫外检测法测定牛奶中青霉素G....... 59 第三部分双三元液相色谱-在线固相萃取在工业产品中的应用. .62 DGLC-I-A01在线固相萃取-高效液相色谱-紫外检测法测定纺织品中直链烷基苯磺酸盐(LAS)). 63DGLC-I-A02在线固相萃取-高效液相色谱-紫外检测法测定聚合物中偶氮二异丁腈(AIBN). ....67 第四部分双三元液相色谱-在线固相萃取在药物代谢中的应用.......... 71 DGLC-M-A01在线固相萃取-高效液相色谱-质谱-质谱检测法测定鼠血浆中杀鼠灵.... 72 DGLC-M-A02 在线固相萃取-高效液相色谱-紫外检测法测定人血浆中苯芴醇 ....... 75 DGLC-M-A03 在线固相萃取-高效液相色谱-质谱-贡谱检测法测定人血浆中非索非那定...... ..79 DGLC-M-A04 在线固相萃取-高效液相色谱-紫外检测法测定鼠血浆中氢氯噻嗪和尼群地平 .85 DGLC-M-A05在线固相萃取-高效液相色谱-紫外检测法测定血浆中的抗真菌药 91 第一部分 双三元液相色谱-在线固相萃取在环境领域的应用 DGLC-E-A01L在线固相萃取-高效液相色谱-紫外和荧光检测联用测定自来水中的多环芳烃 关键词:在线固相萃取;高效液相色谱;紫外检测器;荧光检测器;自来水;多环芳烃 DGLC-E-A01 Hydrocarbons (PAHs) in tap water using online solid-phase extraction followed by HPLCwith UV and fluorescencedetections Key words: Online solid-phase; HPLC; UV detector; fluorescence detector; tap water; PAHs 引言 多环芳烃类化合物(PAHs)是强烈的致癌物质,因此它们在食品和环境中的存在受到关注。世界各国的法规都限制了饮用水,食品添加剂,化妆品,工作场所,以及工厂排出物中多环芳烃类化合物的含量。通常使用高效液相色谱法(HPLC)分离测定多环芳烃质类化合物,但是 HPLC 所使用的直接注射样品的方法检出限太高,不适用于检测实际样品中和限制标准相接近的低浓度 PAHs, 因此样品需要在检测前预浓缩。美国EPA 描述用液-液萃取,液-固萃取(也称为固相萃取)2作为样品预浓缩的方法,但该方法费时,费人工,并且每个样品需用新的固相萃取柱也花费不少。相比之下,在线固相萃取 (On-line SPE) 结合HPLC是一-种简单,快速,精确的检测方法。 Dionex UltiMateQ 3000 智能液相色谱系统的设计则为高级液相色谱分析提供了经济有效、精确的方法,完成饮用水样品中美国EPA 优先控制污染物名单所列出的16种多环芳烃类化合物在 EPA 550.1方法所列检测限浓度下的在线固相萃取-HPLC的分析3。 测试条件 仪器: UltiMate 3000 HPLC 系统, 包括 DPG 3600A泵(带脱气机), TCC-3100 柱温箱(带六通阀), WPS 3000TSL自动进样器,VWD-3400RS 可变波长检测器, RF2000 荧光剂监测器,使用变色龙软件控制仪器运行。 分析柱: SUPELCOSILTM LC-PAH (5 um, 4.6 mmx250 mm) 富集柱: Acclaim PA 2 (3 um, 4.6 mmx50 mm) 柱温:20°C 进样体积:2.0mL 淋洗液组成、流速及淋洗梯度条件:见表1。 检测波长:紫外检测254 nm, 荧光检测(多种激发和发射波长编程)见表2。 图1-1 冲洗富集时连接图 图1-2 分析时连接图 图 仪器连接图 表1 系统梯度淋洗条件 时间 (min) 左泵: A-—水;B- 乙腈。 右泵:A- 水;B- 乙。 valve 流速 (mL/min) A% B% curve 流速(mL/min) A% B% curve 0.00 1.00 95 5 1.00 60 40 1_2 8.00 1.00 95 5 5 1.00 60 40 5 6_1 8.50 0.50 0 100 5 1.00 60 40 5 10.00 0.50 0 100 5 1.00 60 40 5 30.00 0.50 0 100 5 1.00 0 100 5 54.00 0.50 0 100 5 1.00 0 100 5 1_2 54.50 1.00 95 5 5 1.00 60 40 5 65.00 1.00 95 5 5 1.00 60 40 5 表2荧光检测波长 时间(min) 激发波长(nm) 发射波长(nm) 增益 0.00 256 390 1 31.50 275 420 4 34.00 270 385 1 37.00 290 430 1 51.00 305 480 4 65.00 256 390 1 样品前处理 结果和讨论 方法的重现性、线性和检出限 在上述色谱条件下进行试验,各PAHs的线性、检出限数据见表3,加标回收率在70%~131%。 表3 PAHs线性、检出限 分析物 线性范围(ug/kg) 线性系数 检测方法 检出限 (ug/L) 萘 1.00~25.00 0.9950 紫外检测法 1.17 苊烯 2.00~50.00 0.9994 紫外检测法 1.08 苊 1.00~25.00 0.9986 紫外检测法 0.84 芴 0.20~5.0 0.9994 紫外检测法 0.11 菲 0.10~2.50 0.9986 荧光检测法 0.15 蒽 0.10~2.50 0.9969 荧光检测法 0.08 荧蒽 0.20~5.00 0.9943 荧光检测法 0.09 芘 0.10~2.50 0.9945 荧光检测法 0.26 苯并(a)蒽 0.10~2.50 0.9843 荧光检测法 0.08 苯并菲 0.10~2.50 0.9952 荧光检测法 0.15 苯并(b)荧蒽 0.20~5.00 0.9964 荧光检测法 0.017 苯并(k)荧蒽 0.10~2.50 0.9991 荧光检测法 0.01 苯并(a)芘 0.10~2.50 0.9982 荧光检测法 0.022 二苯并(a,h)蒽 0.20~5.00 0.9984 荧光检测法 0.025 苯并(g,h,i)二萘嵌苯 0.20~5.00 0.9989 荧光检测法 0.070 茚苯(1,2,3-cd)芘 0.10~2.50 0.9992 荧光测法 0.059 实际样品分析 在自来水样品中加入美国 EPA 优先控制污染物名单所列出的16多环芳烃类化合物标准对照品,连续进样8次,保留时间的 RSD 在 0.03%~0.13%之间,色谱峰面积的 RSD 在1.0%~10.8%之间。 图2.加入多环芳烃标样的自来水的色谱图 (8次连续进样的重叠图) SPE Column:n:Acclaim PA2, 3 pm, 4.6×50 mm Peaks: 1. Naphthalene 5.0 pg/L (A)254 nm 紫外检测(a为空白对照,b为自来水,c为加多环芳烃标准品的自来水) (B)不同波长荧光检测(a为空白对照,b为自来水,c为加多环芳烃标准品的自来水) 图 3.样品色谱图 注意事项 方法的干扰来自溶剂、试剂中的杂质、玻璃器皿及其他的样品处理过程,所以高灵敏度实验必须使用高纯试剂,实验所需器皿必须仔细清洗干净。 在程序中应加入清洗 2.5 mL 样品环路,,以减少残留。 具体程序如下:20.000 WashSampleLoop Volumn=2500.000 22.000 Wash 25.000 InjectValveTolnject 在分析大量样品的时候,建议在进样器和转换阀之间加一个在线过滤器(0.2um),以保护固相萃取柱和分析柱。 结论 (1) 在线SPE (OnLine SPE) 方法对自来水中多环芳烃的测定可完全满足EPA方法550.1的要求。 (2)该方法简单可靠、重现性高而且线性好,节省分析时间,比离线固相萃取节省费用。 (3) 戴安UltiMate 3000系列双梯度泵配合变色龙软件可以简单、方便地实现OnLine SPE的所有要求。 ( 参考文献 ) ( [1] Determination o f Polycyclic Aromatic Hydrocarbons in Drinking Water by Liqu i d-Liquid Extraction and HPLC w i th Coupled Ultraviolet and Fluorescence Detection, Method 550, U.S . EPA ) ( [2] Determination of Polycyclic Aromatic Hydrocarbons in Drinking Water by Liquid-Solid E x traction and HPLC with Coupled Ultraviolet and Fl u orescence Detection, Method 550.1, U.S. EPA. ) ( [3] D ionex Corporation. D e termination of Phenols in Drinking and Bottled Mineral Waters Using Online ) ( Solid-Phase Extraction Followed by HPLC with UV Detection, Application Note 191, LPN 1949-02.Sunnyvale,CA, 2008. ) DGLC-E-A02 在线固相萃取-高效液相色谱-紫外检测法测定饮用水和瓶装矿泉水中苯酚 关键词:在线固相萃取;高效液相色谱法;紫外检测器;饮用水;瓶装矿泉水;苯酚 DGLC-E-A02 Determination of phenols in drinking and bottled mineralwater by HPLC-UV detection with online solid- phase extraction Key words: online solid-phase extraction; HPLC; UV detector; drinking; bottled mineral water;phenol 引言 苯酚类化合物是一种水体污染物,欧盟规定饮用水中每种苯酚类化合物的限量为 0.5 ug/L,日本厚生劳动省(Ministry ofHealth, Labour and Welfare)规定饮用水中苯酚类化合物的含量不得高于5 ug/L2, 美国EPA规定五氯苯酚及优先污染物列表③中的11种苯酚类化合物(11种苯酚类化合物结构式见图1)限量为1ug/L[4],采用 GC 法进行检测。常见的检测方法有GC5,6] ,GC-MS79,因样品中一些物质对 图1 美国EPA公布的优先污染物列表中的11种苯酚类化合物结构图 富集柱: IonPacNG1 Guard (5 um, 4.0 mmx35 mm); 柱温:40℃; 检测波长:280 nm; 进样量:10mL; 淋洗液组成、流速及淋洗梯度条件:见表1。 图2仪器连接图 表1 系统梯度淋洗条件 左泵程序: A=0.2 mmol/L MSA 溶液, B=ACN 右泵程序: A=25 mmol/L HAC 溶液/25 mmol/LNH4Ac 溶液(1.45:1; v/v), B=ACN 时间(min) 流速(mL/min) A% B% 备注 流速(mL/min) A% B% 备注 -14.0 1.0 0 100 冲洗SPE柱 0.2 0 100 冲洗分析柱 -13.0 0.2 75 25 -11.5 1.0 0 100 -11.0 1.0 99 1 平衡SPE柱 -8.6 2.0 99 1 载入样品 -5.0 1.0 75 25 平衡分析柱 -3.5 2.0 99 1 -3.0 1.0 85 15 冲洗SPE柱 0.0 1.0 75 25 进样 0.2 0 0 0 3.5 0.2 0 100 17.5 1.0 30 70 18.0 1.0 0 100 样品前处理 将样品通过0.45 u.m滤膜过滤,取495 mL滤液,加入5mL甲醇和56 uL MSA (最终试液中MSA的浓度约为2 mmol/L),待测。 结果和讨论 流动相种类及浓度对酚保留时间的影响 许多酸溶液110-12]可以作为流动相来分离酚类化合物;本试验对分析色谱条件中的A通道溶液进行了优化,分别将A通道溶液换成0.1 mM 甲基磺酸溶液(液3A)、0.1%三氟乙酸溶液(图3B)、0.1%乙酸溶液(图3C)及25mM 乙酸-乙酸铵(1:1,v/v)缓冲液(图3D),酚类分离情况见图3. 图3酚类化合物(浓度10 pg/mL) 的色谱图 色谱峰: (1). Phenol; (2). 4-Nitrophenol; (3). 2-Chlorophenol; (4). 2-Nitrophenol; (5). 2,4-Dimethylphenol; (6).2,4-Dinitrophenol; (7).4-Chloro-3-methylphenol; (8). 2,4-Dichlorophenol; (9). 4,6-Dinitro-2-methylphenol;(10).2,4,6-Trichlorophenol; (11). Pentachlorophenol。 试验表明,甲基磺酸溶液、三氟乙酸溶液、乙酸溶液对酚类分离的选择性基本一样,而乙酸一乙酸铵缓冲液则不同。我们研究了流动相中酸浓度变化对各种酚类保留值的影响。试验表明,除了某些酚外,大部分组分的保留时间随酸浓度的变化都不明显。当使用MSA溶液时,浓度从 0.1 mmol/L 提高到3 mmol/L 时, 2,4-dinitrophenol的保留时间逐渐缩短,其色谱峰的位置从2,4-dimethylphenol 之后提前到 2-nitrophenol 之前。当使用 HAc 溶液时,浓度从0.03%至2.0%,各个酚的保留时间移动倾向类似于 MSA 溶液。用 TFA 代替 HAc 结果一样。而当使用25 mM 的 HAc-NH4Ac 缓冲体系时,不同的 pH值(酸碱比例不同)使2,4-dinitrophenol及44,6-dinitro-2-methylphenol1的保留时间变化远大于酸溶液。同时2,4,6-trichlorophenol 及 pentachlorophenol 也有很大的变化。 流动相的选择 不管使用HAc、MSA还是TFA溶液,美国EPA604中所指定的所有11个酚都能被很好的分离,酸的浓度更低时分离会更好。但由于酸的浓度很低时其溶液易受环境中二氧化碳的影响导致酸浓度变化,而某些酚对酸的浓度变化又比较敏感;从而进一步导致了方法的重现性变差。因此我们选用HAc-NH4Ac缓冲盐体系作为流动相,在浓度为25mM,并且酸与碱体积之比为:1.45比1时,有更好的分离度及方法重现性。 优化在线SPE方法 我们试验了不同浓度的酸(HAc或MSA)与不同浓度甲醇或乙腈混合,作为SPE的洗脱溶液洗提杂质并保留、浓缩酚类,试验表明,使用酸/乙腈溶液时的效率更高,所以我们使用0.2mmol/L MSA/乙是为提脱液以降低背景。 线性、检出限 在上述色谱条件下,按 EPA Method 604中的线性浓度进行试验。线性、检出限数据见表2。 表2 11中苯酚类化合物线性、检出限 序号 分析物 线性范围(ug/L) 线性系数 检出限 (ug/L) EPA 604检出限(ug/L) GC-FID GC-ECD 1 2,4-二硝基苯酚 0.5~20 0.9998 0.46 13.0 0.63 2 苯酚 0.5~20 0.9984 0.87 0.14 2.2 3 4,6-二硝基-2-甲基苯酚 0.5~20 0.9998 0.40 16.0 未检出 4 4-硝基苯酚 0.5~20 0.9997 0.42 2.8 0.70 5 2-氯苯酚 0.5~20 0.9996 0.41 0.31 0.58 6 2-硝基苯酚 0.5~20 0.9992 0.41 0.45 0.77 7 2,4-二甲基苯酚 0.5~20 0.9999 0.30 0.32 0.68 8 4-氯-3-甲基苯酚 0.5~20 0.9998 0.31 0.36 1.8 9 2,4-二氯苯酚 0.5~20 0.9998 0.08 0.39 未检出 10 2,4,6-三氯苯酚 0.5~20 0.9999 0.20 0.64 0.58 11 氯苯酚 0.5~20 0.9965 0.93 7.4 0.59 注::a——-t检测估算MDL,置信度为99%时, MDL等于一定浓度的标准溶液(2ug/L) 重复测定7次的峰面积标准偏差S与对于的t值。 实际样品的分析 选取瓶装矿泉水、饮用蒸馏水和自来水进行加标回收试验,试验数据见表3~6,色谱图见图 4~7. 1-figure8_1 mineral water 2 add 10uL MS/ 18.( 图4瓶装矿泉水1#加标叠加图 图5并瓶装矿泉水2#加标叠加图 1,瓶装矿泉水1#;2,瓶装矿泉水1#加标) (1,瓶装矿泉水2#;2,瓶装矿泉水2#加标) 1-figure10_110ppb 2-figure10_2tap water add 10 uL MS/ 18.0 图6饮用蒸馏水样品加标叠加图 图7自来水加标叠加图 (1,饮用蒸馏水样品;2,饮用蒸馏水样品加标) (1,标准图谱;2,自来水样;3,自来水加标) 表3.瓶装矿泉水1#加标回收试验 峰序号 化合物名称 测得值 标准加入值 加标测得值 回收率 (ug/L) (ug/L) (ug/L) (%) 1 2,4-二硝基苯酚 未检出 10 9.44 94 2 苯酚 未检出 10 11.9 119 3 4,6-二硝基-2-甲基苯酚 95.6 4 4-硝基苯酚 未检出 10 10.2 102 5 2-氯苯酚 未检出 10 10.4 104 6 2-硝基苯酚 119 7 2,4-二甲基苯酚 未检出 10 10.5 105 8 4-氯-3-甲基苯酚 未检出10 9.56 95.6 9 2,4-二氯苯酚 97.5 10 2,4,6-三氯苯酚 未检出 10 10.1 101 11 五氯苯酚 0.73 10 9.67 96.7 注::样品进样2次,取平均值;加标样品进样4次,取平均值。 表4.瓶装矿泉水2#加标回收试验 峰序号 化合物名称 测得值 标准加入值 加标测得值 回收率 (ug/L) (ug/L) (ug/L) (%) 1 2,4-二硝基苯酚 未检出 10 9.57 95.7 2 3 苯酚 0.37 10 10.0 9.57 100 95.7 4 4,6-二硝基-2-甲基苯酚 4-硝基苯酚 未检出 10 未检出 10 10.0 100 5 2-氯苯酚 未检出 10 9.02 90.2 6 2-硝基苯酚 未检出 10 10.9 109 7 2,4-二甲基苯酚 未检出 10 9.97 99.7 8 4-氯-3-甲基苯酚 未检出 10 9.40 94.0 9 2,4-二氯苯酚 未检出 10 9.05 90.5 10 2,4,6-三氯苯酚 0.75 10 9.55 95.5 11 五氯苯酚 未检出 10 9.60 96.0 注:样品进样2次,取平均值;加标样品进样5次,取平均值。 表5.饮用蒸馏水加标回收试验 峰序号 化合物名称 测得值 标准加入值 加标测得值 回收率 (ug/L) (ug/L) (ug/L) (%) 1 2,4-二硝基苯酚 4.95 99.0 2 苯酚 未检出 54.84 5.02 96.8 3 4,6-二硝基-2-甲基苯酚 未检出 55.02 100 4 4-硝基苯酚 5.09 102 5 2-氯苯酚 未检出 55.22 104 6 2-硝基苯酚 未检出 5 5.30 106 7 2,4-二甲基苯酚 8 4-氯-3-甲基苯酚 未检出 5 5.07 101 9 2,4-二氯苯酚 未检出 5 4.98 99.6 10 2,4,6-三氯苯酚 未检出 5 5.20 104 11 五氯苯酚 未检出 5 4.99 99.8 注::样品进样5次,取平均值;加标样品进样4次,取平均值。 表6.自来水水加标回收试验 峰序号 化合物名称 测得值 标准加入值 加标测得值 回收率 (ug/L) (ug/L) (ug/L) (%) 1 2,4-二硝基苯酚 2.11 15 10.4 70.0 2 苯酚 0.41 15 14.2 94.7 3 4,6-二硝基-2-甲基苯酚 未检出 15 15.1 101 4 4-基基苯酚 0.80 15 15.2 101 5 2-氯苯酚 低于检出限 15 11.50 76.7 6 2-硝基苯酚 未检出 15 14.0 93.3 7 2,4-二甲基苯酚 1.63 15 15.0 100 8 4-氯-3-甲基苯酚 低于检出限 15 14.5 96.4 9 2,4-二氯苯酚 未检出 15 14.1 94.0 10 2,4,6-三氯苯酚 0.65 15 14.6 97.0 11 五氯苯酚 1.13 15 14.2 94.5 注: 样自进样2次,取平均值;加标样品进样5次,取平均值。 使用双梯度泵的其中一个泵作为大体积进样器 如果所需分析的样品量不多,可以不使用自动进样器。而用双梯度泵中的一个泵代替AS-HV。该泵的A及B可放置溶剂,而C可以放置样品。根据泵的流量及时间确定进样量。 注意事项 方法的干扰来自溶剂、试剂中的杂质、玻璃器皿及其他样品的处理过程,所以高灵敏度实验必须使用高纯试剂,实验所需器皿必须仔细清洗干净。 大体积进样时,样品的 pH 值需用 MSA 调整至3.5以下。 AS-HV 自动进样器的管路及定量环与高有机含量的溶剂不兼容,因此必须用不锈钢或PEEK 材料对相应的部件或管路进行改造。 结论 (1)在线SPE (OnLine SPE) 方法对环境中酚的测定可完全满足EPA方法604的要求。 (2)该方法简单可靠、重现性高而且线性好,灵敏度达达GC的水平,而且适应性更好。 (3)戴安UltiMate 3000系列双梯度泵配合变色龙软件可以简单、方便地实现OnLineSPE的所有要求。 ( 参考文献 ) ( [ 1 ]. D rinking Water Directive 80/778/EEC, Commission of the European Communities, 1980. ) ( [2]. M inistry Ordinance No. 15,M i nistry of Health and Welfare, T okyo, J apan, 2000. ) ( [3] j . . U.S . E nvironmental P rotection Agency. Current N ational Recommended d W ater Quality C ri teria. http://www.epa.gov/waterscience/criteria/wqcriteria.html (accessed Aug 23,2007) ) ( [4]. U.S.EPA Title 40, Chapter 1, Part 1 41, National Primary Drinking Water Regulation. ) ( [5]. F i amegos, Y.C.; Na n os, C.G.; Pilidis, G.A.; Stalikas, C.D. Phase-Transfer Catalytic Determination of Phenols as Methylated Derivatives by Gas Chromatography with Flame Io n ization and Mass- Selective Detection. J.Chromatogr.,A 2003,983,215-223. ) [6]. U.S. Environmental Protection Agency. 40 CFR 136: Appendix A to Part 136, Methods for OrganicChemical Analysis of Municipal and Industrial Wastewater, Method 604—Phenols. Cincinnati, OH,1984 ( [7]. P eng, X .; W ang, Z ; . Y a ng, C.; C h en, F. ; M a i , B. S imultaneous Det e rmination of E ndocrine-Disrupting Phenols and Steroid Estrogens in Sediment by Gas Chromatography-Mass S p ectrometry. J. Chromatogr.,A 2006,1116,51-56. ) ( [8]. M ontero, L.; C o nradi, S . ; Weiss, H . ; P o pp, P . Determination of Phenols in La k e and Ground Water Sampl e sby Stir Bar S orptive Extraction-Thermal Desorption-Ga i s s Chromatography-Mass Spectrometry.J. Chromatogr.,A 2005,1071,163-169. ) ( [9]. S araji, M.; Bakhshi, M . Determination o f Phenols in Water S a mples by Si n gle-Drop Mi c roextraction Followed by In-Syringe e D erivatization andGas Chromatography-Mass Spectrometric > D etection. J.Chromatogr., A 2005, 1098,30-36. ) ( [ 1 0]. Application Update 119, Phenols, Dionex Corporation, 2000 ) ( [1 1] . Acclaim Column Brochure, D ionex Corporation, p34, 2006 ) [12]. D. Xuan, Y. Li, China Public Health, 18 (2002) 1102 DGLC-E-A03 在线固相萃取-高效液相色谱-紫外检测法测定水样中痕量微囊藻毒素 关键词:在线固相萃取;高效液相色谱法;紫外检测器;水;微囊藻毒素 DGLC-E-A03 Determination of trace microcystins in water byHPLC-UV detection with online solid-phase extraction Key words: online solid-phase extraction; HPLC; UV detector; water; microcystins 引言 微囊藻毒素 (Microcystins,MCs)是水体中有害蓝藻水华释放出来的一类具有强致癌作用的肝毒素,迄今已发现60多种异构体。已知存在的最普遍、含量相对较多、毒性较大的主要是 MC-LR,MC-RR, MC-YR三种(见图1)。微囊藻毒素有很高的耐热性,加热煮沸都不能将毒素破坏,也不能将其去除。自来水处理工艺的混凝沉淀、过滤、加氯也不能将其去除。目前,我国《生活饮用水卫生标准》(GB/T 5749-2006)1规定了饮用 MC-LR R=CH(CH3)2 MC-RR R=CH2CH2NHC(NHz)NH MC-YR R=C6H,OH 图1 微囊藻毒素结构图 水中的 MC-LR 含量不得高于1 ug/L。世界卫生组织(WHO)在其推荐的饮用水标准指导(第二版,1998)2中增加了微囊藻毒素等指标,要求 MC-LR 含量不得高于1 ug/L。 国家标准 GB/T 20466-2006《水中微囊藻毒素的测定》③中的高效液相色谱法采用离线样品处理,将过滤后的1L水样通过固相萃取柱进行富集,经洗脱、浓缩后测定,检出限为0.1ug/L。该方法操作繁琐,处理试样量大,给实验带来不便。本方法利用双三元液相系统,实现在线富集,10 mL 进样量即可达到检出限0.1ug/L 的要求,操作简单,灵敏度高,试验成本低。 测试条件 仪器: Ultimate DGP 3600系列,包括有带在线脱气单元的双三元梯度泵;配有2500 uL 半制备进样组件的自动进样器;带有一个六通阀的柱温箱;紫外检测器。连接图见图2。 分析柱: AcclaimPAII (3 um, 4.6 mmx150 mm) 富集柱: AcclaimPAIIGuard Cartridge(5 um, 4.3 mmx10 mm) 柱温:30℃ 检测波长:238 nm 进样量:10mL 淋洗液组成及流速:i:见表1 梯度淋洗:见表2 图2仪器连接图 表1 淋洗液组成及流速 表2 梯度淋洗条件 富集泵(left pump) 分析泵(right pump) 时间(min) 富集泵(B%) 时间(min) 分析泵(B%) 时间(min) 阀 流动相A 20mM KHPO4缓冲溶液(用50% H;PO4 调pH至 2.5) 0 20 0 20 0 1-2 流动相B MeOH ACN 4 20 7 20 7 6-1 流速 2.0 mL/min 1.0 mL/min 10 50 15 50 11 1-2 11 70 17 50 16 70 18 70 样品前处理方法 将样品通过0.45 um滤膜过滤,置于10mL样品瓶中,待测。 图3 标准溶液色谱图(各离子浓度均为5 ug/L) 结果和讨论 用户自定义进样程序 (User Defined Program, UDP) 变色龙软件具有强大的用户自定义功能,其UDP 进样功能可实现不同样品瓶内溶液混合、扩展进样体积等操作。本实验使用的进样组件为半制备进样组件,一次最大进样量可达2.5 mL。但由于所测目标物在样品中含量极低,需更大体积进样量才能达到检测要求。通过UDP功能,可实现连续多次进样以达到大体积富集的效果。 上样流动相选择 试验对比了用纯水和20 mM KHPO4(pH2.5)缓冲溶液作为富集泵的流动相A的区别。当用纯水时,按照上述色谱条件进行分析,发现自来水样品加标测定时, MC-YR 和 MC-LR峰丢失,加标样品中只有 MC-RR 峰且回收率良好。原因可能是自来水样品中的组分干扰了富集柱对 MC-YR 和 MC-LR的保留,通过更换不同填料富集柱,减少进样体积,增加进样时对富集柱的洗脱时间和洗脱强度均无改善。最终,将富集泵的流动相A 更换为 20 mMKH2PO4(pH2.5)缓冲溶液,解决了 MC-YR 和 MC-LR 峰丢失现象。 线性、检出限 按上述色谱条件,以峰面积进行评价,本测定方法在0.5 ug/L~ 10 pg/L 浓度范围内线性良好,三种微囊藻毒素的线性相关系数 R> 0.9996,检出限(S/N=3)为 0.1 ug/L。 实际样品分析 选取自来水和湖水进行加标回收试验,扣除空白,收试验数据见表3~5,色谱图见图4~5。从试验数据可以看出, MC-RR 的加标回收率在 100%~112%之间; MC-YR的加标回收率在100%~107%之间; MC-LR的加标回收率在92%~102%之间,方法准确性良好。 图4 自来水加标叠加图 图5 湖水加标叠加图 (1, [自来水加标扣空白;2,自来水样) (1,湖水加标;2,湖水样) 表3 MC-RR 加标回收试验 样品名 测得值 标准添加值 加标测得值 加标回收率 (ug/L) (ug/L) (ug/L) (%) 自来水 未检出 11.048 104.8 未检出 10 10.36 103.6 湖水 未检出 1 1.116 111.6 未检出 10 10.38 103.8 表4MC-YR 加标回收式验 样品名 测得值 标准添加值 加标测得值 加标回收率 (pg/L) (ug/L) (ug/L) (%) 自来水 未检出 1 1.061 106.1 未检出 10 10.06 100.6 湖水 未检出 1 1.011 101.0 未检出 10 10.21 102.1 表5MC-LR加标回收试验 样品名 测得值 标准添加值 加标测得值 加标回收率 (ug/L) (ug/L) (ug/L) (%) 自来水 未检出 10.923 92.3 未检出 10 10.04 100.4 湖水 未检出 1 0.973 97.3 未检出 10 10.13 101.3 结论 该方法测定时间短,回收率高,样品前处理简单,所需样品量少,可满足现行相关法规对水体中3种常见痕量微囊藻毒素的同时检测。 ( 参考文献 ) ( [1]. 中华人民共和国国家标准, GB/T 5749-2006《生活饮用水卫生标准》 ) ( [2 ] . G uidelines for Drinking Water Quality , 2nd Ed ) ( [3]. 中华人民共和国国家标准, GB/T 20466-2006《水中微囊藻毒素的测定》 ) DGLC-E-A04 在线固相萃取-高效液相色谱-紫外检测法测定环境水样中四种痕量邻苯二甲酸酯 关键词:在线固相萃取;高效液相色谱法;紫外检测器;水;痕量;邻苯二甲酸酯 DGLC-E-A04 Determination of four trace phthalate esters in environmentalwater by HPLC-UV detection with online solid-phase extraction Key words: online solid-phase extraction; HPLC; UV detector; environmental water; trace;phthalate ester 引言 邻苯二甲酸酯是一种增塑剂,被广泛应用于化工、医药、食品等各个领域。塑料及其制品中的邻苯二甲酸酯在一定的环境下,迁移至与之接触的环境或食品中,最终通过呼吸道或食物链进入人体,造成内分泌紊乱。此外,男性睾丸癌和生殖器官发育不良也与这种化学物质有关。我国生活饮用水卫生标准 GB 5749-2006《生活饮用水卫生标准》中规定水质中DEHP 的含量不得高于8ug/L。 目前对于邻苯二甲酸酯的测定有气相色谱-质谱联用法和液相色谱法,如 GB/T 22048-2008《玩具及儿童用品聚氯乙烯塑料中邻苯二甲酸酯增塑剂的测定》[2 GB/T21911-2008《食品中邻苯二甲酸酯的 图1 四种邻苯二甲酸酯结构图 测定》3和 HJ/T 72-2001《水质邻苯二甲酸二甲(二丁、二辛)酯的测定液相色谱法》4,《水和废水监测分析方法(第四版)》中也分别介绍了液相色谱法和 GC-MS 法检测水体中邻苯二甲酸酯。食品及环境水样中的邻苯二甲酸酯的含量很少,传统的样品处理操作繁琐,有机溶剂消耗大,处理样品量大,给试验带来不便。本方法利用双三元液相系统,实现在线富集,各项指标符合国标要求,操作简单,灵敏度高,试验成本低。 带有一个六通阀的柱温箱;紫外检测器。连接图见图2。 分析柱: Acclaim 120 C18 (5 um, 4.6 mmx150 mm) 富集柱: IonPac"NG1 Guard (10 um, 4.0 mmx35 mm) 柱温:40℃ 检测波长:224 nm 进样量:15mL 淋洗液组成及流速!::见表1 梯度淋洗::见表2 图2 仪器连接图 表1 淋洗液组成及流速 表2 梯度淋洗条件 富集泵(left pump) 分析泵 (right pump) 用于上样,流速1.0 流动相A HO mL/min 流动相B ACN 流速 1.0 mL/min 时间(min) 分析泵(B%) 阀 备注 -15 30 1-2 富集柱富集,分析柱平衡 0 30 6-1 富集结束,触发阀切换 1.0 30 6-1 开始信号采集 2.0 30 6-1 梯度洗脱 6.0 100 6-1 13.0 100 6-1 13.1 10 6-1 分析结束,平衡富集柱 15.0 10 6-1 分析完成 样品前处理 将样品通过0.45 um滤膜过滤,待测。 图3 标准溶液色谱图(待测物浓度均为 10 ug/L) 结果和讨论 IonPacNG1 Guard 柱连接方式 对于富集泵液体与分析泵液体流经 IonPacNG1 Guard 柱的方向是否相同,延伸出两种连接方式,一种是富集泵液体与分析泵液体流经IonPacNG1 Guard 柱的方向相反(连接图见图2),即“反向冲洗”,可有效防止色谱峰扩散;另一种是富集泵液体与分析泵液体流经IonPac NG1 Guard 柱的方向相同(连接图见图4),即“同向冲洗”,这种连接利用富集柱的分离作用进行初步的分离提纯。本试验对比了两种连接对试验结果的影响,在其他条件均相同的情况下,当采用同向冲洗连接方式时,色谱峰明显变宽拖尾,当采用反向冲洗连接方式时,色谱峰型尖锐且对称性好。故采用反向冲洗连接。 图4-1 阀1-2相通时连接图 图4-2 阀1-6相通时连接图 图4 “同向冲洗”时连接图 色谱条件的优化 当梯度淋洗有机相(ACN)起始梯度为50%时, DMP 色谱峰在梯度峰上,影响定量测定。通过优化,将有机相起始梯度降低为30%,,再延缓有机相比例至100%的时间,获得良好的效果。 线性、检出限 按上述色谱条件,以峰面积进行评价,本测定方法在0.5 ug/L~ 50 ug/L 浓度范围内,四种邻苯二甲酸酯线性良好,线性相关系数 R>0.9991, 检测限(按S/N=3) DMP 为 0.1 ug/L,DEP 为 0.25 ug/L, DBP 为 0.10 ug/L, DEHP 为 0.25 ug/L。 实际样品的分析 选取自来水和湖水进行加标回收试验,试验数据见表3~4,色谱图见图5~6。从试验数据可以看出, DMP 的加标回收率在 95%~101%之间, DEP 的加标回收率在 95%~102%之间,DBP 的加标回收率在 93%~102%之间, DEHP 的加标回收率在 88%~108%之间,方法准确性良好。 图5自来水加标叠加图 图6湖水加标叠加图 (a,自来水样; b,自来水加标) (a,湖水样;b,湖水加标) 表3 自来水中四种邻苯二甲酸酯加标回收试验 待测物 测得值 标准添加值 加标测得值 加标回收率 (ug/L) (ug/L) (pg/L) (%) DMP 未检出 5 4.760 95.2 未检出 50 49.157 98.3 DEP 未检出 5 4.911 98.2 未检出 50 48.107 96.2 DBP 未检出 5 5.071 101 未检出 50 48.363 96.7 DEHP 未检出 5 5.070 101 未检出 50 49.757 99.5 表4 湖水中四种邻苯二甲酸酯加标回收试验 待测物 测得值 标准添加值 加标测得值 加标回收率(ug/L) (ug/L) (ug/L) (%) DMP 未检出 5 5.040 101 未检出 50 49.307 98.6 DEP 未检出 5 5.051 101 未检出 50 47.907 95.8 DBP 未检出 5 4.971 99.4 未检出 50 46.903 93.8 DEHP 2.44 5 6.856 88.3 2.44 5056.153 107 结论 该方法测定时间短,回收率高,样品前处理简单,所需样品量少,可满足现行相关法规对水体中四种痕量邻苯二甲酸酯的同时检测。 ( 参考文献 ) ( [1]. . 中华人民共和国国家标准, GB/T 5749-2006《生活饮用水卫生标准》 ) ( [2]. 中华人民共和国国家标准, GB/T 22048-2008《玩具及儿童用品聚氯乙烯塑料中邻 苯 二甲酸酯增塑剂的测定》 ) ( [3]. 中华人民共和国国家标准, GB/T 21911-2008《食品中邻苯 二 甲酸酯的测定》 ) ( [4]. 中华人民共和国环境保护行业标准, HJ/T72-2001《水质邻 苯 二甲酸 二 甲( 二 丁、 二 辛)酯的测定液相色谱法》 ) ( [5 ] .王心芳.《水和废水监测分析方法(第四版)》.北京:中国环境科学出版社,2002 ) DGLC-E-A05 在线固取萃取-高效液相色谱-荧光检测法测定污水处理厂水样中直链烷基苯磺酸盐(LAS) 关键词:在线固相萃取;高效液相色谱;荧光检测法;污水;直链烷基苯磺酸盐 DGLC-E-A05 Determination of Linear Alkylbenzene Sulphonate intreatment plant wastewater streams using online solid-phaseextraction followed by HPLC with fluorescence detection Key words: online solid-phase extraction; HPLC; fluorescence detection; wastewater; linearalkylbenzene sulphonate 引言 表面活性剂为洗涤剂主要活性成分,而直链烷基苯磺酸盐(LAS, CAS No.68411-30-3)则是其中使用较为频繁的一种阴离子表面活性剂,市售产品一般是其同系物及异构体的混合物1-9], LAS结构参见图1. 近来来出现了数种测定环境样品中LAS 的分析方法,主要应用在农业土壤和沉积物分析方面。这些方法大多是基于液相色谱,检测器为 DAD(二极管阵列检测器)、FLD(荧光检测器)或MS(质谱)。直接注射样品的方法检出限较高,不适合自 图1 LAS 结构图 然水体检测,若要检测则需在分析前需要水样进行富集。固相萃取(SPE) 技术是最重要的样品富集技术之一,它克服了许多液液萃取的缺点,但其单个样品处理较费时,且 SPE 小柱往往仅能使用一次。而在线固相萃取技术结合高效液相色谱检测的方法则可节约成本,减少手工操作、是一种简便、快速、准确的分析方法。 本文利用双三元高效液相色谱系统(DGLC-3600),采用在线固相萃取方式(Online SPE)以IonPac"NG1 小柱对样品进行在线富集,后转载到分析柱 AcclaimSurfactant 上进行分离。Chromeleon 软件可以很容易地控制该在线固相萃取高效液相色谱方法所用双三元硬件。与其它分析方法相比,本方法可节省时间及成本、避免接触大量有机溶济,显著提高分析效率。 测试条件 仪器型号: Ultimate DGP 3600 系统,包含::带有在线脱气单元的双三元梯度泵,配有 500 uL 半制备进样组件的自动进样器,带有一个二位十通阀的柱温箱,荧光检测器,系统连接参见图2. 分析柱: Acclaim Surfactant (5 um, 4.6 mmx 250 mm);保护柱: Acclaim Surfactant Guard(4.3mm ×10 mm) 富集柱: IonPac NG1(5 um, 4 mmx 35 mm) 检测器:荧光检测器,0 min: Ex 230 nm, Em 302 nm; 18 min: Ex 221 nm, Em 284 nm柱温:30℃ 进样量:0.5-5mL 淋洗液组成及流速i::见表 1 表1 淋洗液组成及流速 富集泵(left pump) 分析泵(B%) 分析泵(right pump) 阀位置 流动相 HO 流动相A ACN 20 15 流动相B 100 mmol/L NH4OAC (pH 5HCl调节) 流速 2.0 mL/min 流速 1.0 mL/min 图2仪器连接图 样品前处理 取污水样品适量,过滤,并尽快测试以防止水样变质。 结果和讨论 在线固相萃取色谱条件优化 通过进不同量的 C10-13 LAS 混标来考察方法的线性,谱图见图3,线性数据见表3 图3不同进样体积 C10-13 LAS 色谱图 (LAS标准品总浓度 58.5 ug/L, a-0.5 mL; b-1.0 mL; c-2.0 mL; d-3.0mL; e-5.0 mL) 重现性、检出限和线性 由于样品中所含 LAS 的浓度范围较宽,所以本实验的方法建立了 0.5mL、5mL两个不同进样量的标准工作曲线,并分别测得了两个进样量的重现性(表4)、检出限(表5)和工作曲线线性情况(表3)。 表3线性数据 SPE 进样量(mL) C10r C11r C12r C13r 0.5 0.9967 0.9964 0.9962 0.9935 5 0.9996 0.9988 0.9988 0.9975 表4重复性数据 SPE 进样 C10 C11 C12 C13 RT Area Are Area Area 量(mL) RT RSD RT RSD RT RSD RSD RSD RSD RSD RSD 0.5 (n=5) 0.24 1.72 0.22 1.33 0.16 1.39 0.12 3.25 5 (n=3) 0.95 11.1 0.03 9.9 0.04 15.1 0.03 15.4 表5 LAS检出限 SPE 进样 C10 C11 C12 C13 量(mL) LOD (ug/L) RSD LOD (ug/L) RSD LOD (ug/L) RSD LOD (ug/L) RSD 0.5(n=5) 5.1 3.29 14.2 3.89 15.4 1.1 12.3 3.86 5 (n=3) 0.19 11.4 0.51 12.1 0.46 12.4 0.31 15.3 实际样品的分析 研究了污水处理厂入水口及出水口中 LAS 总量,进样量0.5mL, 谱图见图4、图5。本实验也对 LAS 含量较低的样品进行了研究,进样5mL,谱图见图6、图7。 图4某城市污水及工业污水处理厂入水 口污水样品谱图(LAS总量为8689pg/L) 图5某城市污水及工业污水处理厂出水口污水样品谱图 (LAS 总量为740pg/L) 图6某污水处理厂出水口污水样品谱图(LAS 总量为 37.1ug/L) 图7某污水处理厂出水口低LAS含量污水样品加标117ug/L谱图(实线为样品 谱图,虚线为加标样谱图) 加标回收率 低含量样品添加 117 ug/L 标准品 C10-13 LAS 以研究加标回收率,图7中对比了低含量样品及其加标样品的谱图,样品平均加标回收率为 104%, RSD 为 14%(n=3)。 结论 Ultimate 3000 双三元系统结合 Online SPE方法可成功用于水样中 LAS的分析,而无需进行手工离线样品前处理。Online SPE 结合荧光检测器检测,可以获得良好的重复性和选择性。5 mL 样品时 LAS C10-13总量的检出限为 0.9 ug/L 注意事项 严格以高纯水清洗所有玻璃器皿,并使用高纯度试剂和溶剂,以尽量减少干扰问题。样品必须过滤并在短时间进行分析,以免待测物降解。 ( 参考文献 ) ( [ 1] . H ERA,2004. Human and Environmental Risk A ssessment of Ingredients of Household Cleaning Products:Report for LAS. h ttp://www.heraproject.com. ) [2]. Mieure, J.P.; Waters, J.; Holt, M.S.; Matthijs, E. Terrestrial Safety Assessment of Linear AlkylbenzeneSulphonate Chemosphere 1990,21,251-262. [3]. Kloepper-Sams, P.; Torfs, F.; Feijtel, T.C.J.F.; Gooch, J. Effects Assessment for Surfactants inSludge-Amended Soils: a Literature Review and Perspective for Terrestrial Risk Assessment. Sci. Tot.Environ. 1996,185,171-175. [4]..De Wolf,W.; Feijtel, T.C.J.F. Terrestrial Risk Assessment for Linear Alkylbenzene Sulfonate (LAS) inSludge-Amended Soils. Chemosphere 1998, 36 (6), 1319-1343. ( [5]. Jensen, J. Fate and Effects of Linear Alkylbenzene Sulphonates (LAS) in t he T errestrial Environment-A Review Sci. Tot. Environ. 1990, 226,93-111. ) ( [6]. . J ensen, J.; Lokke,H.; Holmstrup,M.; Krogh, P.H . ; Elsgaard, L. Effects and Risk Assessment of LinearAlkylbenzene Sulfonates in Agricultural Soil . Probabilistic Risk Assessment of Linear AlkylbenzeneSulfonates in Sludge-Manded Soils. Environ . Toxicol. Chem. 2001,2 0 (8) , 1690 - 1697. ) ( [7]. F endinger, N.J.; Versteeg, D . J.; Weeg, E.; Dyer, S.; R a paport, R.A. Environmental Behavior and Fate of Anionic S urfactants. In:Baker LA, editor. Environmental Chemistry of Lakes and Re s ervoirs. ACSAdvances in Chemistry S eries American Chemical Society 1994,528-57. ) [8]. Versteeg, D.J.; Rawlings,J.M. Bioconcentration and Toxicity of Dodecylbenzene Sulfonate (C12LAS) toAquatic Organisms Exposed in Experimental Streams. Arch. Environ. Contam. Toxicol. 2003, 44, 237-46. [9].TTemara, A.; Carr, G.; Webb,S.; Versteeg, D.J.; Feijtel, T. Marine Risk Assesment: LinearAlkylbenzensulphonates (LAS) in North Sea. Marine Pollution Bulletin 2001 42(8),635-42. DGLC-E-A06在线固相萃取-高效液相色谱-紫外检测法测定污水处理厂水样中直链烷基苯磺酸盐(LAS) 关键词:在线固相萃取;高效液相色谱;紫外检测;废水;直链烷基苯磺酸盐 DGLC-E-A06 Determination of Linear Alkylbenzene Sulphonate intreatment plant wastewater streams using online solid-phaseextraction followed by HPLC with UV detection Key words: online solid-phase extraction; HPLC; UV detection; wastewater; linear alkylbenzenesulphonate 直链十二烷基苯磺酸盐 (LAS)是合成洗涤剂中含有的重要阴离子表面活性剂之一。LAS 进入水体后,与其它污染物结合形成分散胶体颗粒,对工业污水和生活污水的物理、化学和生化特性都有很大影响,并产生潜在的危险。 GB 8978-1996 《中华人民共和国国家标准污水综合排放标准》规定LAS一级排放限量为5 mg/L"。 目前测定污水中 LAS 的方法主要有流动注射法和分光光度法。国标(GB/T 7494-1987)采用亚甲蓝分光光度法21,操作麻烦,基体干扰大,灵敏度低(试样体积100mL时,方法检出限为 0.05 mg/L)。流动注射法是改进的亚甲蓝方法的自动化版本,用仪器法代替人工的有机溶剂萃取,但仍可能存在较大的基体干扰,易出现假阳性。本文的方法无需多次萃取,利用在线前处理小柱 NG1 可以有效地纯化、富集LAS, 大大提高检测灵敏度。戴安公司的 Surfactant 表面活性剂色谱柱可以对不同碳链的 LAS 进行分离和测定, 7.5mL进样时检出限为 1ug/L. 测试条件 仪器型号: Ultimate DGP 3600 系统,包括有带在线脱气单元的双三元梯度泵;配有2.5mL 半制备进样组件的自动进样器;带一个二位六通阀的柱温箱; DAD 检测器。连接图见图2. 分析柱: Acclaim Surfactant (5 um, 4.6 mmx250mm) 保护柱:Acclaim Surfactant Guard (4.3mm x10 mm) 富集柱: IonPac NG1(5 um, 4 mmx 35 mm) 柱温:30℃ 表1 淋洗液组成及流速 富集泵(Left pump) 分析泵 (Right pump) 流动相A ACN ACN: 100 mmol/L NH40AC= 流动相B HO (7:3, v/v) (pH=5.0, HCl调节) 流速 2 .0 mL/min 流速 1.0 mL/min 表2 梯度淋洗条件 时间 富集泵 时间 阀位置 (min) (B%) (min) 0 95 0 1-2 10 95 3 6-1 11 20 8 1-2 20 20 25 95 35 95 2-1 阀1-2位相通时连接图 图2-2 阀1-6位相通时连接图 图2 仪器连接图 样品前处理 取污水样品适量,过滤,并尽快测试以防止水样变质。 结果和讨论 标准品色谱图 图3标样色谱图(LAS总浓度为100 ug/L) 检出限和线性 按上述色谱条件,以峰面积进行评价,本测定方法在50ug/L~ 1 000 ug/L 浓度范围内,LAS线性良好,相关系数r>0.9995。以3倍噪音的峰高为检出限,以 50 ug/L 的谱图为参考标准谱图,获得混标谱图中 LAS-C11 的峰高和基线噪音,计算得到该方法最低检出限为lug/L. 实际样品的分析 图4为某污水处理厂入水口水样及其加标样品色谱图重叠图。图5为某污水处理厂出水口水样及其加标样品色谱图重叠图。 图4某污水处理厂入水口水样及其加标样品色谱图重叠图 图5某污水处理厂出水口水样及其加标样品色谱图重叠图 (1-加标图;2-入口水样) (1-加标图;2-出口水样) 加标回收率 称量样品时加入适当体积的标准溶液(1.0 mg/L 和10 mg/L),以得到加标 100 ug/L 和500 ug/L 的样品。两个污水处理厂的出、入口水样的加标回叫率实验结果见表3。 表3 LAS 加标回收实验数据 样品名称 沙得值 (ug/L) 标准添加值(ug/L) 加标测得值(ug/L) 回收率(%) 污水处理厂1# 入口出口 11.35 100 93.92 82.6 13.82 100 95.82 82.0 污水处理厂厂2# 入口 出口 57.7 500 478.2 84.1 22.32 500 481.8 91.9 结论 本方法适合检测污水处理厂污水中的直链十二烷基苯磺酸盐(LAS),基体干扰少,回收率高。7.5 mL 进样, LAS 总量检出限为 1ug/L,灵敏度高于国标方法。 利用戴安公司双三元液相色谱系统 (Ultimate 3000 DGLC),在线固相萃取技术 (OnlineSPE),实现样品在线富集和基体去除,,可以实现对低含量样品的准确测定。用作SPE 柱的IonPac NG1,兼容100%水相上样,可反复使用,节约检测成本。 注意事项 严格以高纯水清洗所有玻璃器皿,使用高纯试剂和溶剂,以尽量减少干扰问题。样品必须过滤并在短时间进行分析,以免降解。 ( 参考文献 ) ( 中华人民共和国国标标准 GB 8978-1996 《中华人民共和国国家标准污水综合排放标准》 ) ( 中华人民共和国国标标准 GB/T 7494-1987《水质阴离子表面活性剂的测定亚甲蓝分光光度法国家标 准》 ) DGLC-E-A07在线固相萃取-高效液相色谱-紫外检测法测定水样中痕量莠去津(阿特拉津) 关键词:在线固相萃取;高效液相色谱;紫外检测;环境水;莠去津 DGLC-E-A07 Determination of trace levels of Atrazine inenvironmental water using online solid-phase extraction followed byHPLC with UV detection Key words: on-line solid-phase extraction; HPLC; UV detection; environmental water; Atrazine 引言 莠去津(Atrazine)是一种三嗪类除草剂,又名阿特拉津,结构式见图1。已知莠去津是一种内分泌干扰物,莠去津污染是一个普遍而又危险的环境问题。 《生活饮用水卫生标准》(GB 5749-2006)中规定水质中莠去津的限量为0.002 mg/L"。常规HPLC检测时,需对样品进行有机溶剂液液萃取或者大体积离 图1 莠去津结构图 线固相萃取,前处理较复杂,且所需样品量较大。本方法利用双三元高效液相系统 (DGLC-3600),采用在线固相萃取方式(Online SPE)对样品进行富集后再进行HPLC检测。 进样2.5mL时,检出限可达0.05 ug/L。该方法显著减少样品使用量,实现自动化样品在线前处理和测定,, 节省分析时间、避免使用大量有机溶剂,可大大提高分析效率。 测试条件 仪器: Ultimate DGP 3600 系统,包括有带在线脱气单元的双三元梯度泵,配2.5mL半制备组件的自动进样器,带有一个六通阀的柱温箱,紫外检测器,系统连接参见图2。 分析柱: Acclaim 120 C18 (5um, 4.6mmx250mm) 富集柱: Acclaim"PA Cartridge(5um,4.3mmx10mm) 柱温:30℃ 表1 淋洗液组成及流速 检测波长:222nm 进样量:2.5mL淋洗液组成及流速::见表1: 梯度淋洗条件:见表2。 富集泵(Left Pump) 分析泵 (Right Pump) 流动相A MeOH 流动相A MeOH 流动相B HO 流动相B HO 流速 1 mL/min 流速 1mL/min 表2 梯度淋洗条件 时间(min) 富集泵(B%) 时间(min) 分析泵(B%) 时间(min) 阀位置 0 100 0 30 0 100 15 30 6 9.5 0 15.1 0 7 1-2 18 0 22 0 _ 18.5 100 22.1 30 23 100 30 30 图2-1 阀1-2位相通时连接图 图2-2 阀1-6位相通时连接图 图2 仪器连接图 样品前处理 将水样通过0.22 um尼龙滤膜过滤,滤液待测。 结果和讨论 标准品色谱图 图3标准溶液色谱图(浓度10ug/L) 线性、检出限 以0.5、1、2、4、6、8、10 pg/L 7个浓度的标样色谱峰面积对浓度做工作曲线,具有良好线性,线性相关系数 r 大于0.9998。以3倍信噪比计算检出限,得到该方法最低检出限为 0.05 ug/L 实际样品的分析 (1)自来水及其加标1ug/L、2ug/L、5ug/L样品的重叠谱图见图4,加标回收率见表3。 图4自来水样品及样品加标对比谱图 (1-自来水;2-加标1 ug/L;3-加标2 ug/L;4-加标5 ug/L) (2)河水及其加标 1ug/L、2ug/L、5ug/L样品的重叠谱图见图5,加标回收率见表3。 图5河水样品及样品加标对比谱图 (1-河水;2-加标1 ug/L; 3-加标2 pg/L;4-加标5 ug/L) 表3 莠去津的加标回收实验结果 样品名称 原样含量(ug/g) 加标量(ug/g) 测得含量(ug/g) 加标回收率(%) — 1.003 1.045 104.2 自来水 2.005 2.038 101.6 5.006 5.028 100.4 1.002 1.091 108.9 河水 2.004 1.971 98.4 5.001 4.971 99.4 结论 本方法利用双三元液相色谱系统的在线固相萃取技术,实现样品在线富集和基体消除,避免繁琐的人工样品前处理过程,大大节省样品使用量和时间,重现性高,回收率好。 本实验使用半制备进样组件,一次最大进样量可达2.5 mL, 结合SPE柱的富集功能,可以实现对低浓度样品的准确测定。水样2.5mL进样,检出限可达0.05 ug/L, 灵敏度高,适合水体中莠去津的检测。 ( 参考文献 ) ( [1]. 中华人民共和国国家标准, GB 5749-2006《生活饮用水卫生标准》 ) 第二部分 双三元液相色谱-在线固相萃取在食品饮料中的应用 DGLC-F-A01 在线固相萃取-高效液色色谱-荧光检测法测定食用油中多环芳烃 关键词:在线固相萃取!;;高效液相色谱法;荧光检测器;食用油2;;多环芳烃 DGLC-F-A01 Determination of Polycyclic Aromatic Hydrocarbons(PAHs) in Edible Oils by HPLC-Fluorescence detection with onlinesolid-phase extraction Key words: online solid-phase extraction; HPLC; Fluorescence detector; edible oil; PAHs 引言 多环芳烃类化合物(PAHs)是强烈的致癌物质,而食用油在其制造过程中会产生一定的多环芳芳类物质。因为苯并芘(benzo[a]pyrene) 是多环芳烃中具有代表性的化合物,欧盟规定了食用油中的苯并芘含量不得高于 2.0 ug/kg. 多环芳烃质类化合物的测定,通常使用高效液相色谱法(HPLC)分离,采用紫外检测器[2]、荧光检测器[3.4]、电化学检测器以及质谱6测定,经氧化反应后,还可采用 LC-MS/MSI进行检测,因常规方法中样品前处理采用离线固相萃取,手工操作繁琐,使其重现性受到影响。虽然已经有文献报道了在线固相萃取法分析多环芳烃类化合物,但装置极其复杂。本方法利用双三元液相系统,一套系统即可实现在线富集,大大加快了试验过程,操作简单,灵敏度高,试验成本低。 测试条件 仪器: Ultimate DGP 3600系列,包括有带在线脱气单元的双三元梯度泵;带有两个六通阀的柱温箱;荧光检测器;自动进样器。连接图见图2。 分析柱:2根SUPELCOSILTMLC-PAH (5 um, 4.6 mmx250 mm)富集柱: ChromSpher Pi (3.0 mmx80 mm) 柱温:40℃;柱温:30℃ 自动进样器温度:40℃ 进样量:80pL 见表1。检测波长:见表2。 图1-2 冲洗富集时连接图 图1-3分析时连接图 图1 仪器连接图 表1 系统梯度淋洗条件 时间(min) 左泵 A-水;B-乙腈;C-异丙醇 left valve 右泵 A-乙腈;B-异丙醇 rightvalve 流速(mL/min) A% B% C% 流速(mL/min) A% B% 0.00 0.35 0 0 100 6_1 0.4 20 80 1_2 12.00 0.35 0 0 100 12.10 0.35 20 80 0 61 14.50 1_2 14.60 0.4 20 80 16.00 1.0 20 80 17.00 6_1 20.90 0.35 20 80 0 20.91 0.35 0 100 0 30.00 1.0 0 100 50.90 0.35 0 100 0 51.50 0.35 0 0 100 58.00 1.0 0 100 58.10 1.0 20 80 61.50 12 65.00 1.0 20 80 65.50 0.4 20 80 66.50 0.35 0 0 100 70.00 0.4 20 80 表2 检测波长 时间(min) 激发波长(nm) 发射波长(nm) 0.00 256 370 27.05 256 390 29.50 240 420 33.50 270 385 37.50 290 430 51.50 305 480 53.50 290 430 样品前处理方法 将食用油试样过 0.45 um 滤膜,移取 40 uL 0.25 ug/mL 苯并(b)屈内标物至 10g食用油试样中,混匀待测。 结果和讨论 方法的重现性 在橄榄油样品加入美国 EPA 610 方法所列出的12种多环芳烃类化合物标准对照品,连续进样7次,保留时间的 RSD 在0.03%~0.08%之间,色谱峰面积的 RSD 在 4.1%~6.7%之间。 线性、检出限 在上述色谱条件下进行试验,各PAHs的线性、检出限数据见表3。 表3PAHs线性、检出限 分析物 线性范围(ug/kg) 内标浓度(ug/kg) 线性系数 检出限 (ug/L) 菲 1~20 1 0.9952 0.42 蒽 1~20 1 0.9910 0.26 荧蒽 1~20 1 0.9808 1.19 芘 1~20 1 0.9905 0.69 苯并(a)蒽 1~20 1 0.9852 0.68 屈 1~20 1 0.9864 0.34 苯并(b)荧蒽 1~20 1 0.9907 0.21 苯并(k)荧蒽 1~20 1 0.9907 0.39 苯并(a)芘 1~20 1 0.9911 0.75 二苯并(a, h)蒽 1~20 1 0.9914 0.41 苯并(g,h,i)二萘嵌苯 1~20 1 0.9920 0.58 茚苯(1,2,3-cd)芘 1~20 1 0.9941 0.59 注:a-t检测估算MDL,置信度为99%时, MDL等于一定浓度的标准溶液 (2ug/kg) 重复测定7次的峰面积标准偏差S与对于的t值。 表 4食用油中多环芳烃含量的测定及加标回收试验 PAH 橄榄油1# 橄榄油2# 芝麻油 测得值 (ug/kg) 标准加入值(ug/kg) 回收率(%) 测得值 (ug/kg) 测得值(ug/kg) 菲 37 5 120 13.2 52 蒽 4.5 5 109 3.2 6.1 荧蒽 1.0 5 112 未检出 未检出 芘 2.2 5 131 1.3 未检出 苯并(a)蒽 2.8 5 108 2.1 18 屈 4.4 5 110 3.2 5.3 苯并(b)荧蒽 未检出 5 90 未检出 未检出 苯并(k)荧蒽 未检出 5 84 未检出 未检出 苯并(a)芘 2.7 5 106 2.5 3.9 二苯并(a, h)蒽 未检出 5 84 未检出 未检出 苯并(g,h,i)二萘嵌苯 未检出 5 70 未检出 1.2 茚苯(1,2,3-cd)芘 未检出 5 82 未检出 未检出 结论 本方法使用在线固相萃取方法对食用油中多环芳烃进行测定,操作简便,减少手动操作步骤,大大提高了方法的重现性。 ( 参考文献 ) ( [1 ] . C ommission Regulation (EC) No . 18 8 1/2006 of 19 Dec e mber 2006 , Sett i ng Maximum Levels for Certain Contaminants in F o odstuffs. Official Journal of the European U nion 2006, 49, L364,5-24. ) ( [2]. S aravanabhavan, G.; Helferty, A.; Hodson, P. V;Brown, R . S. A Multi-Dimensional H igh Performance L i quid Chromatographic Method for Fi n gerprinting Polycyclic Aromatic Hydrocarbons and Their Alkyl-Homologs in the Heavy Gas Oil Fraction of Alaskan North Slope C r ude. J. Chromatogr., A 2007, 115 6 , 124-133. ) [3]. Zuin, V. G.; Montero, L.; Bauer, C.; Popp, P. Stir Bar Sorptive Extraction and High-Performance LiquidChromatography-Fluorescence Detection for the Determination of Polycyclic Aromatic Hydrocarbons inMate Teas. J. Chromatogr., A 2005,1091,2-10. ( [4]. P ino, V.; A yala, J. H.; Afonso, A.M.; G o nzalez, V. D etermination of P olycyclic Aromatic Hydrocarbons in Seawater t by H H igh-Performance L Liquid Chromatography yW with F l uorescence D etection i F lollowing Micelle-Mediated Preconcentration. J.Chromatogr., A 2002, 949,291-299 ) [5]. Bouvrette, P.; Hrapovic, S.; Male, K. B.; Luong, J. H.Analysis of the 16 Environmental Protection AgencyPriority Polycyclic Aromatic Hydrocarbons by High Performance Liquid Chromatography-OxidizedDiamond Film Electrodes.J. Chromatogr., A 2006,1103,248-256. ( [6]. I toh, N .; A oyagi, Y.; Y arita, T . Optimization o f the Dopant fo r the Trace De t ermination of P olycyclic Aromatic Hydrocarbons by y L iquid Chromatography/ Dopant-Assisted Atmospheric-Pressure Photoionization/Mass S p ectrometry. J . Chromatogr., A 2006,1131, 285-288. ) ( [7 ] . L intelmann,J.; Fischer, K.; M atuschek, G.Determination of Oxygenated Polycyclic Aromatic Hydrocarbonsin Particulat e Matte r Using High- Performance L iquid Chromatography-Tandem Mas s Spec t rometry. J. Chromatogr., A 2006,1133,241-247. ) DGLC-F-A02 在线固相萃取-高效液相色谱-紫外检测法测定饮料中维生素B12 关键词:在线固相萃取;高效液相色谱法;紫外检测器;饮料;维生素B12 DGLC-F-A02 Determination of cyanocobalamin (Vitamin B12) inbeverages by HPLC-UV detection with online solid-phase extraction Key words: online solid-phase extraction; HPLC; UV detector; beverages; vitamin B12 维生素B12是B族维生素中的一种,不存在于植物中,但鱼、蛋、肉、肝中含量丰富,如果严重缺乏,将导致恶性贫血。如摄入过量,也会对人体产生危害。目前市场上一些功能性饮料中添加了维生素 B12, 添加量层次不齐,相关人群需斟酌饮用。GB/T5009-217《保健食品中维生素B12的测定》中对于饮料样品,取样20 mL 采用免疫亲合法进行富集、萃取处理,检出限3.0 ug/L,处理过程费时耗力。本方法利用双三元液相系统,实现在线富集,大大减少了手工操作,操作简单,灵敏度高,试验成本低。 图1维生素B12结构图 测试条件 仪器: Ultimate DGP 3600系列,包括有带在线脱气单元的双三元梯度泵;配有2500 uL半制备进样组件的自动进样器;带有两个六通阀的柱温箱;紫外检测器。连接图见图2。 分析柱: Acclaim PAII (3 um,3.0 mmx150 mm) 富集柱: Acclaim"PAI (3 um, 4.6 mmx50 mm) 柱温:25℃ 检测波长:361nm 进样量:2.5mL 淋洗液组成及流速:i:见表1; 梯度淋洗:见表2。 UV detector 图2 仪器连接图 表1 淋洗液组成及流速 left pump right pump 流动相 A 25mM磷酸盐缓冲液(称取 3.4gKH PO4 溶于 1L水,用磷酸调 pH至3.2) 流动相B ACN 流速 富集时:1mL/min;冲洗时:1.5 mL/min 0.5mL/min 表2梯度淋洗条件 时间(min) Left Pump (for online SPE) Right Pump (for separation) LeftValve RightValve 流速(mL/min) A% B% 流速 (mL/min) A% B% 0.0 1.0 97 3.0 0.5 90 10 6-1 1-2 7.0 97 3.0 11.4 90 10 11.65 6-1 12.0 70 30 12.15 1.5 1-2 12.5 0 100 14.0 0 100 14.1 70 30 16.0 55 45 16.5 0 100 19.0 70 30 19.1 90 10 19.5 1.0 70 30 20.0 97 3.0 样品前处理方法 将样品通过0.45 u.m滤膜过滤,待测。 图3 标准溶液色谱图(浓度0.5pg/L) 结果和讨论 进样方式 经试验,发现当直接大体积定量环进样时, 实际样品中的杂质成分对维生素 B12色谱峰干扰很大;;当采用传统的在线富集模式进样时,可以去除大多数杂质,但仍无法满足定量的要求。试验在传统的在线富集连接模式的基础上进行了改进(见图2),整个过程见表2,在系统中增加了两个阀,使前期条件摸索阶段,只需通过阀切换即可实现对富集柱和分析柱色谱条件的优化。 线性、检出限 按上述色谱条件,以峰面积进行评价,本测定方法在 0.2 ug/L~ 20 ug/L 浓度范围内,维生素B12,线性相关系数 R>0.9999, 检出限0.003 ug/L。 实际样品分析 选取三种饮料进行加标标收试验,试验数据见表3。从试验数据可以看出,维生素B12回收率在100%~107%,方法准确性良好。 图4 饮料加标叠加图(a,饮料;b,饮料加标) 表3方法测定的回收率 测得值 标准添加值 加标测得值 加标回收率 (ug/L) (ug/L) (ug/L) (%) 1# 未检出 1.5 1.6 107 2# 未检出 2.0 2.1 105 结论 该方法利用双三元液相系统,实现在线富集,大大减少了手工操作,操作简单,灵敏度高,,证试验成本低。 ( 参考文献 ) [1]. 中华人民共和国国家标准, GB/T5009-217《保健食品中维生素 B12的测定》 DGLC-F-A03 加速溶剂萃取-在线固相萃取-高效液相色谱-荧光检测法快速测定谷物或食品中的黄曲酶霉毒素 关键词:加速溶剂萃取;在线固相萃取;高效液相色谱;荧光检测;谷物;食品;黄曲霉毒素 DGLC-F-A03 Fast determination of Aflatoxins in grains or foodusing accelerated solvent extraction follow by online solid-phaseextraction-HPLC with fluorescence detection Key words: ASE; online solid-phase extraction; HPLC; fluorescence detection; grains; food;aflatoxins 引言 黄曲霉毒素由生长在土壤和腐烂植被中的黄曲霉菌所产生,是强致癌毒素。未加工食品中黄曲霉毒素 B1、B2,G1、G2(见图1)的含量在许多国家都受到监管,例如欧盟对谷物中 B1的限量为2.0 ug/kg~8.0 pg/kg, 对黄曲霉素总量的限量为4.0 ug/kg~15.0 ug/kg.美国 FDA 对谷物及食品中的黄曲霉素总量的限量为20ug/kg2。 对于谷物中黄曲霉毒素的检测,常规方法为先进行索氏提取,提取液经离线 SPE 净化后进行 HPLC分离检测,该方法操作繁琐,费时费力。 图1 黄曲霉素B1、B2、G1、G2结构图 本方法采用 ASE、Online SPE-HPLC 方法,可节省时间和人力,提高样品通量,本方法线性、准确度、精度、回收率都很理想。检测限小于2 ppb, 符合欧盟、USFDA要求。 测试条件 仪器型号: Dionex ASE 200; Ultimate DGP 3600 系统,包括有带在线脱气单元的双三元梯度泵;自动进样器;带有一个六通阀的柱温箱;衍生装置; FLD检测器。连接图见图2。 分析柱: Acclaim 120 C18(3um, 4.6x150mm) ( 富集柱: V entureTM AF SPE immunoaffinity ( 15-20um, 2.1mm× 5 0 mm) ) 进样量:5uL 淋洗液组成及流速::见表1; 梯度及阀切换见表2。 表1 淋洗液组成及流速 富集泵(Left pump) 分析泵 (Right pump) 流动相A 10 mmol/L磷酸盐+0.15 mol/LNaCl 混合缓冲盐溶液, pH=7 20%ACN+80% HzO 流动相B HO 22.5/22.5/55 MeOH/ACN/HO 流速 1.0 mL/min 流速 1.0 mL/min 表2 梯度淋洗条件 时间(min) 阀位置 富集泵(%B) 分析泵(%B) 0 1_2 0 0 5.0 1_2 0 0 5.1 12 100 0 10.0 61 100 0 10.1 61 0 0 14.5 1 0 0 14.6 0 100 27.6 0 100 27.7 0 0 40.0 0 图2-1 阀1-2位相通时连接图 图2-2 阀1-6位相通时连接图 图2 仪器连接图 SPE柱 VentureTM AF SPE immunoaffinity column 的固定相选择性的保留目标分析物黄曲霉毒素,富集后的分析物被反冲转载至 Acclaim 120 C18 柱上进行分离。分离之后, B1、 G1 以紫外254nm光化学衍生,转化示意图见图3,从而可用荧光检测器检测这两种黄曲霉毒素。而光化学衍生对B2、G2无作用。 图3 B1在紫外光254nm照射下的转化示意图 样品前处理方法 在ASE200上加速溶剂萃取:称取5 g样品与3g硅藻土混合研磨以除去样品中的水分,并装填萃取池以节省溶剂使用量。处理后的混合物装入22 mL提取池,80℃温度、高压下以体积比50/50的醇醇/乙腈提取5 min, 重复提取两次,样品提取溶剂共16.5mL。而后氮气吹扫120s以除去样品中残留的溶剂。 结果和讨论 标准品色谱图 本实验开发了一种全自动进行样品除杂及检测的 SPE-LC方法,图4是标样色谱图。 400- 图4黄曲霉毒素标样色谱图 (发射光信号, G1、B1浓度 20 ppb, G2、B2浓度6ppb, 进样5pL) SPE 柱上的回收率 本实验通过对比标准品在分析柱单柱以及在分析柱和 SPE 柱双柱上的出峰情况,研究了黄曲霉毒素在免疫亲和 SPE 柱上的回收率(见表3) 表3黄曲霉毒素的回收率(n=2) 黄曲霉毒素 直接进样峰面积(mAU*min) 经SPE柱峰面积 回收率(%) (mAU*min) G2 35.58 38.71 108.8 G1 46.67 44 94.3 B2 52.54 54 102.8 B1 72.80 72.8 100.0 线性、精度及检出限 本实验在黄曲霉毒素浓度 0~20 ug/L 范围内做标准工作曲线,线性、精度及检出限(S/N=5)见表4。精度测定缩使用的混标含有 20 ppb 的 G1、B1以及 4.5 ppb 的 G2、B2。本方法线性、精度良好,检出限满足欧洲立法的规定(食品中B1的限量为2.0 ug/kg,黄曲霉素总量的限量为 4.0 ug/kg)。 表4线性、精度及检出限(n=4) 化合物 回归系数(R2) 保留时间精度 峰面积精度 检出限(ppb) (%RSD) (%RSD) G2 0.997 0.11 1.28 0.6 G1 0.996 0.15 1.34 2.0 B2 0.996 0.14 1.31 0.6 B1 0.996 0.22 1.52 2.0 实际样品的分析 杏仁和玉米样品经 ASE 萃取后, 进行 SPE-HPLC 分析检测,样品谱图见图5,样品保留时间和峰面积精度与标准品相同。 图5样品谱图(左为杏仁;右为玉米) 结论 ASE提取联合Online SPE-HPLC分析谷物和食品中黄曲霉毒素的方法,可节省时间和人力,提高样品通量,本方法在有效浓度范围内线性、精度、选择性和回收率都很理想。本方法可满足相关法规对食品中黄曲霉毒素的检测要求。 ( 参考文献 ) ( [1]. C ommission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Level for Certain C o ntaminants in F o odstuffs. O f ficial Journal of the European Union 2006, 49, L364, 5-24 . ) [2]. Action Levels for Poisonous or Deleterious Substances in Human Food and Animal Feed. Industry ActivitiesStaff Booklet. U. S. Food and Drug Administration, Washington, DC, 2000.http://www.cfsan.fda.gov/~Ird/fdaact.html(accessed Apr30, 2008). DGLC-F-A04 在线固相萃取-高效液相色谱-荧光检测法测定白兰地中的多环芳烃 关键词:在线固相萃取;高效液相色谱;荧光检测;;白兰地;多环芳烃 DGLC-F-A04 Quantification of PAHs in Cognac by HPLC-Fluorescencedetection with online solid-phase extraction with on-line SPE Key words: on-line SPE; HPLC; Fluorescence detection ; Cognac; PAHs 引言 多环芳烃 (polycyclic aromatic hydrocarbon, PAHs)是一类具有多个苯环的碳氢化合物,包括萘、蒽、菲、芘等200余种。此类化合物广泛存在于环境中,他们是由煤、石油、木材、烟草、有机高分子化合物等有机物不完全燃烧时产生的,也可通过原材料、工艺过程带入食品中。大量实验研究证明该类化合物具有强的致癌性。因此,检测环境、食品中的多环芳烃对于保护人类的健康有重要意义。 目前,测定多环芳烃的方法主要有 HPLC 法, 如 EPA 8310", EPA 550.121, HJ 478-2009;GC 法, 如 EPA 8100, EPA 8275A 等。无论哪种方法,都要进行复杂的前处理,处理程序如下:取样品1L,液液萃取并采用硅胶或弗洛里硅土净化;或者采用 SPE 离线净化。不仅操作复杂,而且影响方法的精密度和准确度。 为解决上述方法缺陷,戴安公司利用双三元高效液相色谱仪提供了在线富集、净化及检测的多环芳烃测定的解决方案3。 测试条件 仪器: Ultimate DGP3600系列,包括有带在线脱气单元的双三元梯度泵;酉配有2500 uL半制备进样组件的自动进样器;带有两个六通阀的柱温箱;荧光检测器。 分析柱: Supelcosil LC-PAH, 2.1x250 mm(Supelco P/N 57945) 富集柱: ChromSpher Pi, 3.0x 80 mm (Varian P/N CP28159) 柱温:27℃ 检测波长:见表3 进样量::1mL 淋洗液组成及流速I:: 见表1; 梯度淋洗条件:见表2。 表1 淋洗液组成及流速 left pump(分析泵) right pump(富集泵) 流动相A 40%甲醇 流动相B 乙腈 流动相C 纯水 表2杉梯度淋洗条件 时间 (min) Left Pump(分析泵) Right Pump (富集泵) 左阀 右阀 流速 (mL/min) A% B% C% 流速 (mL/min) A% B% C% 0.0 0.4 0 60 40 0.35 100 0 0 1-2 10-1 15 100 0 0 1-2 15.1 0 60 40 17.8 10-1 23.5 1-2 23.6 0 60 40 25 0 60 40 25.1 0 100 0 10-1 35 0 100 0 35.1 100 0 0 36 0.20 48.6 0 100 0 82.6 0 100 0 85 85.6 0 60 40 0.35 86 92.6 0 60 40 100 0 0 表31RF2000波长切换程序 时间(min) 激发波长(nm) 检测波长(nm) 0.00 300 390 41.00 300 480 42.45 300 425 50.98 300 480 52.30 300 425 76.80 300 480 92.6 300 480 样品前处理方法 样品过微孔滤膜,待测。标准品用40%乙醇稀释。 结果与讨论 进样方式 进样的基本过程如下:首先将样品注入到 DACC* SPE 柱,用适当的淋洗液冲洗,去除基体和杂质的同时将多环芳烃富集在 DACC SPE 柱上(图1A)。待净化完毕,经过阀切换,将 DACC SPE 柱上富集的多环芳烃用分析柱的条件冲洗到多环芳烃专用分析柱上(图1B),然后再次切换回A的状态进行分离后用荧光检测器检测。戴安公司采用上述方案对白兰地中多环芳烃进行了测定,取得了良好的效果。 DACC*: donor-acceptor complex chromatography,一种可与多环芳烃选择性结合并同时除去样品中基质的特殊前处理固相萃取(SPE)柱。 图1A 图1B 图1仪器连接图 使用本方法,可以在93分钟内完成白兰地中13中多环芳烃的分析,并且整个过程完全自动进行,无需任何人工进行的前处理步骤(图2,图3)。但当多环芳烃为ng/L级别,基体中某些成分对检测的影响仍不能完全消除(见图3),有待进一步完善。 图2标准溶液色谱图(80ng/L) 图3样品加标色谱图 线性范围 取标准溶液梯度稀释成五个浓度,每个浓度进两针,以峰高(mV)与浓度(ng/L) 做线性回归。结果显示本方法线性关系良好,线性范围在10-160ng/L 之间,相关系数大于 0.9879(见表4)。 表4标准曲线参数 峰名 相关系数% 截距 斜率 Benzo(a)anthracene 99.89 8.52 0.41 Chrysene 99.97 13.25 0.10 5-Methylchrysene 99.83 2.04 0.12 Benzo(b)fluoranthene 99.89 5.87 0.37 Benzo(k)fluoranthene 99.96 13.65 2.44 Benzo(a)pyrene 99.95 16.04 1.17 Dibenzo(a,l)pyrene 99.97 -0.11 0.73 Dibenzo(a,h)anthracene 99.94 -0.18 0.72 Benzo(g,h,i)perylene 99.95 22.18 0.75 Indeno(1,2,3-cd)pyrene 99.87 0.53 0.08 Dibenzo(a,e)pyrene 99.96 3.00 0.43 Dibenzo(a,i)pyrene 99.89 -0.99 0.12 Dibenzo(a,h)pyrene 98.79 -5.71 0.13 重复性 采用标准加入法进行重复性试验,14种多环芳烃的 RSD 在 0.24%-5.54%之间,结果显示重复性良好(见表5) 表5重复性结果 峰名 加标量 峰面积RSD Benzo(a)anthracene 177.1* 1.84% Chrysene 196.4* 0.06% 5-Methylchrysene 98.8 1.96% Benzo(j)fluoranthene 未检测 Benzo(b)fluoranthene 119.9 2.68% Benzo(k)fluoranthene 115.0 0.31% Benzo(a)pyrene 80.5 1.67% Dibenzo(a,l)pyrene 64.8 0.42% Dibenzo(a,h)anthracene 75.2 0.44% Benzo(g,h,i)perylene 50.8 1.05% Indeno(1,2,3-cd)pyrene 69.9 5.45% Dibenzo(a,e)pyrene 46.1 0.24% Dibenzo(a,i)pyrene 55.5 0.57% Dibenzo(a,h)pyrene 153.3 2.14% 结论 本方法省去了液液萃取,离线 SPE等繁杂操作,实现了在线固相萃取,基体消除和样品分析的全自动化,排除人为误差,获得更精密、准确的结果。 本方法实现 online SPE-HPLC 的关键在于 Dionex 公司的双三元梯度液相色谱仪。该色谱仪的两个梯度泵,并且可以安装双阀。两个泵和两个阀之间可以实现完全的联动操作,因此可以实现一个泵用于固相萃取,另一个泵用于分析的复杂功能。 ( 参考文献 ) ( [1 ] . U.S. EPA, Method 8310.0, Determination of Polycyclic Aromatic Hydrocarbons in Ground Water and Wastes. ) [2]. U.S.EPA, Method 550.1, Determination of Polycyclic Aromatic Hydrocarbons in DrinkingWater by Liquid-Solid Extraction and HPLC with Coupled Ultraviolet and FluorescenceDetection. [3]. Dionex Corporation. Application Note 196, Determination ofPolycyclic AromaticHydrocarbons (PAHs) in Edible Oils by Donor-Acceptor Complex Chromatography(DACC)-HPLC with Fluorescence Detection. DGLC-F-A05 在线固相萃取-高效液相色谱-紫外检测法测定牛奶中青霉素G 关键词:在线固相萃取;高效液相色谱;紫外检测;牛奶;i;青霉素G DGLC-F-A05 Quantification of Penicillin G by HPLC-UV detectionin Milk with on-line SPE Key words: HPLC; UV detection; milk; Penicillin G 引言 青霉素G 为广谱类抗生素药物,有较强的抗菌消炎作用,广泛应用于畜禽类动物的呼吸、胃肠道等脏器细菌感染的预防和治疗。但青霉素G 也易于残留于畜禽体内及其相关产品中,如牛奶、肉等。人类食用这类含残留青霉素 G的产品,会破坏健康人体的正常菌落环境,影响人的身体健康。为保护消费者的食品安全,FDA, 欧盟以及我国都规定了动物组织及牛奶中青霉素G的最大允许残留量。 但由于牛奶、畜禽产品等基体复杂,测定此类产品中痕量的青霉素G 时会由于高背景的存在而受到干扰。同时由于复杂的前处理步骤而导致重复性、准确性较差。因此,要获得牛奶中准确的青霉素含量,必须有效除去基质干扰以及简化操作步骤。 戴安公司应用独特的双三元高效液相色谱仪-在线固相萃取技术成功实现了在线基质去除,从而准确测定了牛奶中痕量的青霉素G。 测试条件 仪器: Ultimate DGP 3600系列,包括有带在线脱气单元的双三元梯度泵;配有2500pL半制备进样组件的自动进样器;带有两个六通阀的柱温箱;紫外检测器。 分析柱: Acclaim PA2, 4.6x250 mm(Dionex P/N063199) 富集柱: LiChrospher RP-8 ADS, 25 pm, 4×25 mm (Merck P/N 1502090001) 柱温:30℃ 进样量:1mL 检测波长:191 nm 淋洗液组成及流速::见表1 梯度淋洗条件:见表2 表 1 淋洗液组成及流速 left pump(分析泵) right pump(富集泵) 流动相A 纯水 流动相B 乙 流动相C 0.1%甲酸 表2杉梯度淋洗条件 时间(min) Left Pump(分析泵) Right Pump(富集泵) 左阀 右阀 流速(mL/min) A% B% C% 流速(mL/min) A% B% C% 0.0 1.0 0 6 94 1.0 50 0 50 1-2 10-1 15 50 0 50 1-2 17 10-1 19 0 6 94 20 0 20 80 21 0 20 80 1-2 28 0 50 50 28.1 0 95 5 32 0 95 5 32.1 50 0 50 10-1 34 35 0 60 40 39 0 60 40 40 0 6 94 46 0 6 94 50 0 50 样品前处理方法 取牛奶样品,在14000g下离心15min,取离心后的中间层液体500pL,转移到截留分子量为3000道尔顿的超滤杯中(Microcon Ultracel YM-3; 3,000 MWCO;Millipore),14000转离心90 min。取滤液进样。 最近超滤杯的供应商更新了产品,新的产品为Amicon Ultra。这种新产品可以使离心时间减少到30分钟。并且新产品有更大的规格,例如一次可以离心4mL样品的型号。旧的型号可以完全淘汰。 图1A 图1B 图1仪器连接图 结论 进样时将青霉素G富集在LiChrospher RP-8 ADS SPE柱上(见图1A),待基质去除完毕,经过阀切换,将青霉素G转移到Acclaim PolarAdvantage II分析柱上(见图1B),再切换到状态A进行分离测定。 适当的前处理(大分子截留超滤)结合在线固相萃取技术(RAM 限制性媒介固相萃取柱),可以用紫外检测器就检测到牛奶中低pg/L级别的青霉素G(图2,图3), 在8 ug/L-80ug/L范围内线性关系良好(相关系数99.87%,图4)。实验中发现在前处理中青霉素有被浓缩的现象,因此建议使用类似于青霉素G且容易被检测的化合物来作为内标物(例如邻苯二甲酸)。另外所选择的波长(191 nm)没有特异性,进一步需要使用质谱检测器。 图2样品色谱图图 图3样品加标20ng/L 色谱图 图4标准曲线 第三部分 双三元液相色谱-在线固相萃取在工业产品中的应用 DGLC-I-A01在线固相萃取-高效液相色谱-紫外检测法测定纺织品中直链烷基苯磺酸盐(LAS) 关键词:在线固相萃取;高效液相色谱;紫外检测;纺织品;直链烷基苯磺酸盐 DGLC-I-A01 Determination of linear alkylbenzene sulphonate intextile using online solid-phase extraction followed by HPLC with UVdetection Key words: online solid-phase extraction; HPLC; UV; textile; linear alkylbenzene sulphonate 引言 直链烷基苯磺酸盐(LAS)通常是指由直链烷基碳数从10到13的同系物(homolog)组成的混合物,结构式见图1。 欧盟纺织品生态标准 Eco-Label明确规定LAS 为禁用和限制使用的部分纺织品化学品之.国标 GB/T 23325-2009 检测 LAS 使用昂贵的LC-MS/MS 连用方法,检出限仅为2 mg/kg.而本方法使用双三元高效液相系统 图1 LAS结构图 (DGLC-3600),采用在线固相萃取 (On-line SPE)技术对样品进行在线富集后再进行 HPLC检测,进样100 uL 样品即可达到总含量 0.15mg/kg 的检出限(通过增大进样体积还可以进一步降低检出限)。该方法能够对含不同碳数的 LAS 进行基线分离,因而可使用紫外检测器进行直接检测,而无需使用质谱。该方法可实现样品的自动化前处理,灵敏度高,回收率好,在线 SPE 柱可重复使用,,i可大大节约测试成本。 测试条件 仪器: Ultimate DGP 3600 系统,包括:带在线脱气单元的双三元梯度泵,自动进样器,带有一个六通阀的柱温箱,紫外检测器,,系统连接参见图2。 分析柱:Acclaim" surfactant (5um, 250 mm x 4.6 mm) 保护柱: Acclaim Surfactant Guard (4.3 mmx 10 mm) 富集柱:]IonPac NG1(5 um, 4x35mm) 柱温:30℃ 检测波长:225 nm 进样量:100pL 淋洗液组成及流速:见表1 梯度淋洗:: 见表2. 表1 淋洗液组成及流速 表2 梯度淋洗条件 富集泵(Left pump) 分析泵 (Right pump) 流动相A ACN ACN: 100 mmol/L NHOAC=7:3(v/v)(pH=5,以盐酸调节) 流动相B H20 流速 1.0 mL/min 流速 1. 0mL/min 时间(min) 富集泵 (B%) 阀位置 -5.0 100 1-2 0 100 6-1 5.2 100 1-2 6.0 0 17.0 0 -- 18.0 100 -- 25.0 100 图2-1 阀1-2位相通时连接图 图2-2 阀1-6位相通时连接图 图2 仪器连接图 样品前处理 取代表性样品,剪成5 mmx5 mm的碎片,混合,称取上述样品0.25 g, 准确加入甲醇5 mL,加塞密闭,置于75℃超声波水浴中提取30min, 冷却至室温。提取液用0.22 um尼龙滤膜过滤后,根据需要用甲醇进一步稀释,待测。 结果和讨论 标准品色谱图 图3为LAS混合标准谱图。LAS的线性烷基链一般在C10~C13,,平均碳链是11.6。市售的LAS约含20多种化合物,包含有不同碳链的同系物及不同取代位置的异构体,也因而 C10~C13的色谱峰形并不规则。 图3混合标准溶液色谱图(LAS总浓度为10 pg/mL) 线性、检出限 按上述色谱条件,以 LAS 总峰面积进行评价,本测定方法在5 pg/mL~100 ug/mL 浓度范围内,线性良好,线性相关系数r> 0.9991。以3倍噪音峰高为检出限,该方法检出限为0.15 mg/kg. 1 1 (C)无纺布样品及无纺布加标20 ug/g (D)绸布样品及样品加标20 ng/g 图4样品及加标样品谱图(1-样品;2-样品加标 20 ug/g) 实际样品的分析 棉布、牛仔布、无纺布、绸布样品及样品加标20 ug/g 谱图见图4,加标回收率见表3 表3 样品中 LAS 的加标回收试验(按LAS总量计算) 样品名称 测得值 (ug/g) 加标示(ug/g) 稀释倍数 加标测得值 (ug/g) 回收率(%) 棉布 95.05 10 2 105.18 101.3 牛仔布 52.3 2 63.23 109.3 无纺布 7.78 31.57 118.9 绸布 2.84 25.58 113.7 结论 本方法使用戴安公司双三元液相色元系统 (Ultimate 3000 DGLC) 在线固相萃取技术(online SPE),实现样品在线富集及基体去除。Acclaimsurfactant色谱柱可以实现对含不同碳数的LAS的基线分离,紫外检测器即可直接检测。以IonPac NG1柱做在线SPE柱,可以兼容100%水相,且可反复使用,节约成本。 本方法适于纺织品中十二烷基苯磺酸钠(LAS)的测定,样品干扰少,回收率高,检出限明显低于国标方法。根据测定需要,还可增大进样量以进一步降低检出限。 ( 参考文献 ) ( [1]. 中华人民共和国国家标准, G B /T 23325-2009《 纺 织品表面活性剂的测定线性烷基苯磺酸盐》 ) DGLC-I-A027在线固相萃取-高效液相色谱-紫外检测法测定聚合物中偶氮二异丁腈(AIBN) 关键词:在线固相萃取;高效液相色谱;紫紫外检测;聚合物;偶氮二异丁腈 DGLC-I-A02 Determination of Azodiisobutyronitrile (AIBN) inpolymer using online solid-phase extraction followed by HPLC withUV Detection Key words: online solid-phase extraction; HPLC; UV detection; polymer; Azodiisobutyronitrile 引言 偶氮二异丁腈(ABIN,结构图见图1)常用作醋酸乙烯和丙烯酸酯等聚合或共聚合的引发剂,其加入量直接影响聚合度。AIBN易燃,有毒,遇热分解放出氮气和含-(CH2)2-C-CN基团的有机氰化物,对人体危害较大。 目前测定AIBN含量的方法有极谱法和分光光度法,极谱法使用剧毒物汞,对人体 图1偶禺二异丁腈(AIBN)结构图 危害极大。分光光度法测定聚合物中 AIBN 的含量方法简便,但是样品基质干扰大。 AIBN 自身紫外吸收较弱,分解温度为64℃,室温下缓慢分解,100℃急剧分解。一般在聚合物中含量较低,采用 HPLC-UV 或者高灵敏度的质谱或者蒸发光散射检测器均无法达到要求。由于此聚合物样品中含有不易挥发的 DMSO, 常见的无需加热的浓缩方式(如氮吹、冻干)也也阻。本文利用双三元高效液相系统(DGLC-3600),采用在线固相萃取方式 (On-line SPE) 对样品进行在线富集后再进行 HPLC检测,实验证明此方法可行。 测试条件 仪器: Ultimate DGP 3600系列,包括有带兑线脱气单元的双三元梯度泵;自动进样器WPS3000TSL(配2.5mL半制备进样组件);带有一个六通阀的柱温箱;紫外检测器。连接图见图2。 分析柱: Acclaim 120 C18 (5 um, 4.6 mmx150 mm) 富集柱: Acclaim120 C18(5 um, 4.3 mmx10 mm) 柱温:20℃ 自动进样器温度:20℃ 检测波长:346nm 进样量:2.5mL 流动相组成及流速i::见表1 阀切换情况:1:见表2。 表1 淋洗液组成及流速 表2 阀切换情况 富集泵(left pump) 分析泵 (right pump) 时间 (min)0 阀位置1-2 备注 进样,样品在 SPE 柱上富集 10%MeAC+90%H,O 50%MeAC+50%HO 7 6-1 富集结束,触发阀切换 13 1-2 等度洗脱 图2 仪器连接图 样品前处理 准确称取1.0g聚合物溶液,在约10倍体积的二氯甲烷中沉淀,于4℃下10000 r/min离心10 min。取上清液,常温下氮吹除去二氯甲烷,得到AIBN的二甲基亚砜溶液,加入1mL甲醇,混合均匀,用水稀释至10 mL,于4℃下10000 r/min再次离心10 min, 过滤,取上清液待测。 结果和讨论 标准品色谱图 图3偶氮二异丁腈(AIBN) 5pg/mL 线性、检出限 按上述色谱条件,以峰面积进行评价,本测定方法在 1.0 ug/mL~ 10 ug/mL 浓度范围内,AIBN 线性良好,线性相关系数 R>0.9995,检测限(按S/N=3) 为 0.1 ug/mL。 实际样品的分析 (1)重结晶 AIBN含量测试,用甲醇配成 10 mg/mL 溶液后,用水稀释为 10 ug/mL 溶液,连续三次进样,谱图见图4,重复性数据见表3。 图4重结晶 AIBN 测试谱图 表3重结晶 AIBN 溶液 10pg/mL重复数据(n=3) 样品 保留时间 峰面积 峰高 含量 塔板数 min mAU*min mAU ug/mL (EP) 10-un-1 12.597 1.6345 11.68 9.6722 52283 10-un-2 12.597 1.6321 11.63 9.6577 51883 10-un-3 12.597 1.6331 11.66 9.6640 52016 Average: 12.597 1.633 11.654 9.665 52061 RSD 0.000% 0.075 % 0.204% 0.075% 0.391% (2)聚合物样品及加标样品谱图 按照样品前处理方法法理后进行 HPLC测试,谱图如图4,加标回收结果参见表4。 图5聚合物样品及样品加标谱图(1-聚合物;2-样品加标) 表4 聚合物样品中AIBN 的加标回收实验 样品测试液含量 折合原样含量 加标量 测得加标样含量 加标回收率 (ug/mL) (ug/g) (ug/g) (pg/g) (%) 1.080 10.80 95.20 79.66 75.2% 结论 采用在线固相萃取技术,同时进行了基体去除和待测组分富集,可有效提高此类热不稳定且紫外响应较弱的化合物的检测则敏度,本方法灵敏度高,方法重复性好。 注意事项 本实验难点在于前处理,样品中含有DMSO,如果进样溶液中有DMSO, 势必仍会有一些聚合物溶解其中,所以要尽量除去DMSO。DMSO熔点较高,为18.5℃,所以采用4℃下离心以最大程度上除去DMSO和溶解在其中的聚合物。 本实验中过滤溶液所用滤膜为尼龙滤膜,DMSO对尼龙有一定程度的溶解,为了保护色谱柱,应该用DMSO专用膜。 ( 参考文献 ) ( [1 ] . 在线化工词典,偶氮二异丁 腈 , ht tp ://hello chem.com/xz/xz1/1493yyeee.ht m ) 第四部分 双三元液相色谱-在线固相萃取在代谢中的应用 DGLC-M-01 在线固相萃取-高效液相色谱-质谱-质谱检测法测定鼠血浆中杀鼠灵 关键词:在线固相萃取;;高效液相色谱;质谱-质谱检测;鼠血浆;杀鼠灵 DGLC-M-01 Determination ofWarfariniin rat plasma byHPLC-MS-MS detection with online solid-phase extraction Key words: online solid-phase extraction; HPLC; MS-MS detection; rat plasma; warfarin 引言 杀鼠灵(Warfarin),又名华法令、华法灵、苄丙酮香豆素、酮苄香豆素,为抗凝血型杀鼠剂,在全世界广泛使用。 鼠血浆中杀鼠灵的常规分析方法是全血样品经乙酸乙酯提取后,在C18柱上以甲醇/0.1%乙酸(50+50, v/v)为流动相进行分离,紫外308 nm波长检测,但灵敏度低,方法回收率不高,重现性较差。 本文详细介绍了在线固相萃取(SPE)方法结合HPLC-MS/MS液质联用方法测定鼠血浆中杀鼠灵的含量。血浆样品在Bio Trap MS C18 (4.0mm×x20 mm)柱上富集,之后在Acclaim PA II柱上进行分离。 图1杀鼠灵结构式 测试条件 仪器型号:Ultimate DGP 3600系统,包括有带在线脱气单元的双三元梯度泵;自动进样器;;带有一个六通阀的柱温箱; API 2000 MS/MS。连接图见图2。 分析柱: Acclaim Polar Advantage ⅡI (5um, 4.6mmx150mm); 富集柱: Bio Trap MS C18(4.0mmx20 mm); 柱温:45℃; 自动进样器温度::l15℃: 进样量:10uL; 淋洗液组成及流速:见表1,等度淋洗; 通阀切换时间见表2; 质谱条件:正离子模式,分流比90:10(10%进入MS),杀鼠灵离子对:309.2/162.9 表1淋洗液组成及流速 表2六通阀切换 分析泵(左泵) 富集泵(右泵) 时间(min) 阀位置 描述 0.1%(v/v)甲酸溶液:ACN (35: 65),用氨水调节pH至5.8±0.05 0.1%(v/v)甲酸溶液/4%异丙醇溶液,用甲酸调节pH至2.5 0-1.5 1-2 清洗及萃取 1.5-3.0 6-1 转载及分离 流速 1.0 mL/min 流速 2.0 mL/min 3.0-7.0 1-2 再平衡 图2仪器连接图 结果和讨论 标准品色谱图 总离子流图见图3,100 ppb标准品图见图4。 图3总离子流图 图4100 ppb标准溶液谱图 实际样品的分析 低浓度加标图见图5. 图5低浓度加标图 结论 本方法中每个 SPE 连续两天使用,共进样约200次,方法重现性好。本方法可成功用于检测鼠血浆中杀鼠灵,方法可靠稳定。 DGLC-M-02在线固相萃取-高效液相色谱-紫外检测法测定人血浆 中苯芴醇 关键词:在线固相萃取;高效液相色谱;紫外检测;人血浆;苯芴醇 DGLC-M-02 Determination of Lumefantrine in human blood plasmausing on-line solid-phase extraction by HPLC with UV detection Key words: on-line solid-phase extraction; HPLC; UVdetection; human blood plasma;Lumefantrine 引言 苯芴醇是我国军事医学科学院发明的-一种疗效好、毒性低的新型抗疟药,其可杀灭疟原虫血液裂殖体,主要用于恶性疟和抗药性恶性疟的治疗[1.2]。 苯芴醇的分析方法目前主要有非水滴定法、紫外分光光度法、HPLC和TLC法等。 本文详细介绍了在线固相萃取 (SPE) 方法结合HPLC紫外检测人体血浆中苯芴醇 含量的方法。血浆样品在LiChrospher RP-8 RAM 柱上富集,之后在聚合物基质的IonPac NS 1其其保护柱NG 1上进行分离。此色谱柱 固定相具有较强疏水性,总运行时间为22 min。 图1苯芴醇结构图 测试条件 仪器型型: Ultimate DGP 3600系统,包括有:带在线脱气单元的双三元梯度泵,自动进样器,带有两个二位十通阀的柱温箱,四通道紫外检测器,硬件连接参见图2-1和图2-2;在图2-1中,样品转移到SPE柱上进行浓缩。之后阀切换至图2-2位置,样品从SPE柱转移至保护柱NG1上,之后阀再次切换到图2位置,样品在分析柱NS1上进行分离。 分析柱: Analytical column: IonPac NS1, (10 um, 4 mmx250mm) 保护柱: IonPac NG1 (10 um, 4 mmx35 mm) SPE柱: LiChrospher RP-8 ADS (25 um, 4 mmx25 mm))((Merck P/N 1502090001) 柱温:30℃ 检测波长:335nm 进样量:5-50pL 淋洗液组成及流速:见表1; 梯度淋洗条件:见表2。 表 1 淋洗液组成及流速 分析泵(左泵) 富集泵(右泵) 流动相A 0.05% TFA 水溶液 (v/v) 流动相A 水 流动相B 0.1%TFA 甲醇溶液(v/v) 流动相B MeOH 流动相C MeOH 流速 1.0 mL/min 流速 1.0 mL/min 表2 梯度淋洗条件 时间(min) 富集泵 时间(min) 分析泵(B%) 时间 (min) 阀位置 (B%) (C%) 左阀 右阀 0 5 25 0 95 0 1-2 10-1 6 5 25 22 95 6 1-2 1-2 6.1 60 0 8.4 10-1 1-2 6.5 95 0 9.6 1-2 1-2 11 95 0 17 1-2 10-1 13 5 25 —— 22 5 25 l图2-1 SPE柱上样时的连接图 图2-22样品由SPE柱转移至预柱连接图 图2 仪器连接图 样品前处理 血浆于4℃下储存,取适量血浆样品,于14000 r/min离心10 min, 取上清液过0.45 um滤膜,待测。 苯芴醇标准品溶解于甲醇/乙酸混合溶液(99.8/0.2,v/v)中 结果和讨论 标准品色谱图 取选取高低两个浓度标准品进样,色谱图如下,高浓度10 ug/mL谱图参见图3,低浓度100 ng/mL谱图参见图4。 图3标准溶液色谱图(浓度10ug/mL) 图4标准溶液色谱图(浓度100 ng/mL) 实际样品的分析 血浆样品及加标400 ng/mL的样品谱图参见图5和图6。 图5血浆样品谱图 图6血浆加标色谱图(加标浓度 400 ng/mL) 结论 本方法利用双三元液相色谱系统在线固相萃取技术,可实现样品基质与苯芴醇的分离。人血浆加标样品中可检测出苯苯醇,但在所需的浓度水平不能进行定量。将苯芴醇从SPE柱上洗脱下来的流动相条件(如高比例的甲醇、强酸性)使得后面的色谱分离比较困难。若使用其它选择性的柱子,例如在较高有机相条件下可以有效保留苯芴醇的柱子,则有可能改善分析状况。使用直径较小的分析柱有助于提高灵敏度。 ( 参考文献 ) ( [1].邓蓉仙,滕翁和,仲景星,焦岫卿,王云玲等.抗疟新药本芴醇及亚油酸胶丸制剂,国家发明一等奖, 1990 ) ( [2] . ] . 邓蓉仙.我国近几年抗疟新药研究近展,中国医药工业志1989, 20:372 ) DGLC-M-03在线固相萃取-高效液相色谱-质谱-质谱检测法测定人血浆中非索非那定 关键词:在线固相萃取;高效液相色谱;质谱-质谱检测;血浆;非索非那定 DGLC-M-03 Determination of Fexofenadine in human blood plasmaby HPLC-MS-MS detection with On-Line Solid-Phase Extraction Key words: on-line Solid-Phase Extraction; HPLC; MS-MS detection; human blood plasma;Fexofenadine: 引言 非索非那定(结构式见图11)是一种抗组织胺药物,用于治疗花粉热和类似过敏症状。血浆中非索非那定60-70%的是与血浆蛋白结合的。口服该药物60 mg后,1-3小时内达到血药浓度峰值209 ng/mL。 本文介绍了在线固相萃取(SPE)结合HPLC-MS/MS 测定人体血浆中非索非那定含量。血浆样品在 Bio Trap 500 C18柱上富集,该富集 图1非索非那定结构式 柱具有表面亲水、孔内疏水的特性,对分子量较 大的血浆蛋白没有保留,对小分子药物可以进行在线富集。药物富集之后被转载至 AcclaimC8柱上进行分离。 测试条件 仪器型号: Ultimate DGP 3600 系统。包含带有在线脱气单元的双三元梯度泵;自动进样器;带有一个六通阀的柱温箱; API2000三冲四级杆质谱检测器。仪器连接图见图2-1、图2-1。 表1 淋洗液组成及流速 表2六通阀切换 分析柱: AcclaimC8, 5 um, 50×4.6 mm SPE 柱: Bio Trap 500 C18, 20×4.0mm 柱温:40℃ 自动进样器温度:15℃ 进样量: 20uL 淋洗液组成及流速:i:见表1;等度淋洗; 六通阀切换时间见表2; 质谱条件:正离子模式,分流比:i: 1:10, SRM检测模式;盐酸非索非那定质量离子对: 502.3/262.2,内标物质量离子对:389.2/201.1 图2仪器连接图 样品前处理 取 50 uL内标(盐酸西替利嗪)加入到 1000 uL已添加非索非那定的血浆样品中,涡旋30秒,10000 r/min 离心8min, 取上清液,进样20puL。 结果和讨论 SRM检测谱图 非索非那定SRM 色谱图见图3。包括在线固相萃取,步骤在内每次测试时间约5min。 2.88 图3非索非那定色谱图(489 ng/mL) 非索非那定保留时间约2.9 min, 内标物盐酸西替利嗪与非索非那定保留时间相同,通过质谱的 SRM 模式可以对其进行选择性检测,质谱图见图5、图6. 4663 图5盐酸西替利嗪(内标物) 图6 非索非那定 选择性 (1) 基质的干扰。 本实验选取了6种不同来源空白血浆,图7是LQC标准样品与空白血浆谱图叠加,基线放大图(图8)显示无基质干扰。 图6 LQC标准样品与空白血浆叠加图(1.LQC标准样品;2.空白血浆) 图7 LQC标准样品与空白血浆基线放大图 (1.LQC标准样品;2.空白血浆) (2) 内标物干扰 SRM 模式对非索非那定和内标物选择性良好(见图8)。空白血浆中未检测到非索非那定和内标物。零血浆(只加内标物的血浆)中检测到389/201的色谱图,为内标物盐酸西替利嗪的特征峰。LOQ标准样品中检测到非索非那定和内标物。 图8空白血浆、零血浆和 LOQ 标准样品的质谱图 准确度及精度 为了验证方法适用性,本实验制作了3日内的工作曲线,标准偏差均小于15% (见表3)。含非索非那定 10 ng/mL、30 ng/mL 的 LOQ标品准确度分别为88.77%和 96.84%。 表3非索非那标准曲线数据 实际浓度 测定浓度(ng/mL) 平均值 S.D R.S.D(%) 平均准确 (ng/mL) 第一天 第二天 第三天 (ng/mL) 度(%) 9.78 9.11 8.41 8.80 8.77 0.351 4.002 89.67 29.34 28.6 28.61 28.15 28.45 0.263 0.924 96.97 48.90 49.54 49.34 53.33 50.74 2.248 4.430 103.76 146.71 162.08 166.35 162.88 163.77 2.270 1.386 111.63 244.51 259.55 268.04 251.24 259.61 8.400 3.236 106.18 391.22 416.03 396.85 409.92 407.60 9.798 2.404 104.19 586.82 585.68 613.74 626.43 608.62 20.853 3.426 103.71 782.43 828.07 839.98 838.75 835.60 6.550 0.784 106.80 1173.65 1171.68 1136.60 1152.89 1153.72 17.555 1.522 98.30 1760.47 1693.9 1685.55 1662.05 1680.50 16.515 0.983 95.46 0.9992 0.9986 0.9985 斜率 0.001 0.00094 0.00094 截距 0.00233 0.00283 0.00115 定量限 低含量样样(含10 ng/mL 非索非那定)测定精度为11.21%(符合小于20%的规定),色谱峰信噪比约为10(符合大于5的规定)。因此, 10 ng/mL 可以被定为定量限。 SPE 柱寿命 本实验使用了两个 BioTrap 500 SPE 柱,第三天的工作曲线是在新柱上测定后绘制的。实验表明,新柱与已使用了800次的旧柱效果相同。图10为旧 SPE 柱分析样品的谱图,图11 为使用新柱子测得的谱图。非索非那定与内标物的 SRM 信号比例相同。另外,研究了非索非那定和内标物在分析柱上的保留时间稳定性。结果显示在每次进样20 uL的情况下,SPE柱可以使用800次以上。 图10旧 SPE 柱测得的谱图 图11 新 SPE 柱测得的谱图 结论 本研究使用 UltiMate 3000 双三元液相色谱系统,利用在线固相萃取-高效液相色谱-质谱联用法测定人血浆中非索非那定。 全自动在线固相萃取测定血浆中非索非那定,样品前处理程序大大简化,样品通量高,该方法的准确度、精确度和线性相关系数符合 ICH 规定",非索非那定定量限为 10 ng/mL。 ( 参考文献 ) ( [1]. B ioanalytical Method Validation. F o od and Drug Administration Center for Drug Evaluation and Research (CDER). M ay 2001. ) DGLC-M-04 在线固相萃取-高效液相色谱-紫外检测法测定鼠血浆中氢氯噻嗪和尼群地平 关键词:在线固相萃取;高效液相色谱;紫外检测器;鼠血浆;氢氯噻嗪;尼群地平 DGLC-M-04Determination of Hydrochlorothiazide and Nitrendipinein rat plasma samples with HPLC-UV detection by online solid-phaseextraction Key words: online solid-phase extraction; HPLC; UV detection; rat plasma; Hydrochlorothiazide;Nitrendipine 引言 在用大鼠进行抗高血压联合用药氢氯噻嗪和尼群地平(结构图见图1)药代动力学实验中,每次取血量有限,且血药浓度较低,要求最好可同时测定氢氯噻嗪和尼群地平。 此两种药物同时检测的分析方法报道很少,多数是对两药分别建立分析方法[1,2]。 图1 氢氯噻嗪和尼群地平结构图 原因主要有两个:一,尼群地 平口服吸收存在首过效应,体内血药浓度值低,大约1-50 ng/mL 在这个检测浓度条件下,多采用液质联用技术进行分析,而此两种药物在质谱工作条件下一个是正离子模式,一个是负离子模式,同时检测不方便;二,尼群地平和氢氯噻嗪极性相差较大,同时提取和分析困难较大。 本文主要介绍使用戴安公司 UltiMate 3000 双三元梯度液相色谱系统对尼群地平和氢氯噻嗪同时测定的方法。 测试条件 仪器: Ultimate DGP 3600系列,包括有带在线脱气单元的双三元梯度泵,自动进样器,带有一个六通阀的柱温箱,紫外检测器,仪器连接参见图2。 分析柱: Acclaim 120 C18 (5um, 4.6mmx250mm); 富集柱: CAPCELL MF C8 (4.0mmx10mm) 柱温:30℃ 表1 淋洗液组成及流速 自动进样器温度:20.0℃ 检测波长:氢氯噻嗪271 nm尼群羊平237 nm 进样量:100uL 淋洗液组成及流速:见表1;右泵梯度淋洗条件:见表2. 富集泵 (Left Pump) 分析泵 (Right Pump) 甲酸水溶液pH=3.00 流速1.0 mL/min 流动相A ACN 流动相B 甲酸水溶液,pH=3.00 流速 1.0 mL/min 表2右泵梯度淋洗条件 时间 分析泵(B%) 阀位置 备注 0 80 1-2 进样、富集柱富集,开始信号采集 1.4 80 6-1 阀切换,将氢氯噻嗪转载至分析柱 2.5 80 1-2 阀切换, SPE 柱切回到富集流路 80 1-2 等度洗脱 10 40 1-2 变化流动相比例 10.5 40 6-1 阀切换,将尼群地平转载至分析柱 15 40 1-2 阀切换, SPE 柱切回到富集流路 24 40 1-2 等度洗脱 25 80 1-2 分析柱平衡,为下一针做准备 34 80 1-2 分析完成 图2-1 阀1-2位相通时连接图 图2-2 阀1-6位相通时连接图 图2 仪器连接图 样品前处理 血浆样品于4℃下,10000 r/min高速离心后,取上清液,用0.22um尼龙滤膜过滤,滤液待测。 结果和讨论 色谱柱的选择及色谱条件优化 本实验分别在 CAPCELL MF C8、CAPCELL MF Ph-1 和 CAPCELL MF SCX 三种 SPE上测试了氢氯噻嗪的保留情况, CAPCELL MF C8对其保留最好,同时该柱对尼群地平也能非常好的保留,故选择此柱作为在线 SPE 柱。 在流通阀切换时间的确定上,本实验中两次将 SPE 柱切入分析流路分别转载两个组分,原因是氢氯噻嗪(HCTZ)保留较弱,第一次转载时血浆基体不能完全流出(见图3),当把氢氯噻嗪完全转移至分析柱后马上将 SPE 柱切换到富集流路,使血浆基体完全流出后,再次将 SPE 柱切入分析流路对尼群地平 (NTDP) 进行转载。 图3血浆加标氢氯噻嗪样品在 SPE 柱上流出情况 标准品色谱图 根据优化好的色谱条件,单次进样可同时检测氢氯噻嗪及尼群地平。氢氯噻嗪标准品色谱图如图4-1,检测波长271 nm;尼群地平标准品色谱图如图4-2,检测波长237 nm。 图4-1氢氯噻嗪(3.3 ppm) 图4-2尼群地平(3.3ppm) 线性、检出限 取大鼠血浆适量,逐级稀释,配制同时含有尼群地平和氢氯噻嗪5、10、20、50、100、500、2000 ng/mL 的血浆溶液 300uL, 于4℃下10000 r/min 高速离心,上清液用 0.22 um的微孔尼龙滤膜过滤后,取100 uL 血浆进样,进行 HPLC分析,将测定所得的尼群地平和氢氯噻嗪紫外吸收的峰面积与对应的血药浓度作图,得出尼群地平和氢氯噻嗪血浆中药物浓度的标准曲线,尼群地平和氢氯噻嗪均采用权重 1/C 进行权重分析。尼群地平的标准曲线为A=0.0061C-0.0047, R=0.9991;;氢氯噻嗪的标准曲线为 A=0.0047C+0.0060,R=0.9991。 以空白血浆分别加标 5 ng/mL 氢氯噻嗪和 5 ng/mL 尼莫地平样品谱图为参照,以3倍信噪比计算检出限,血样中药品检出限:氢氯噻嗪2.5 ng/mL, 尼莫地平2ng/mL。增大血浆进样量,可以检测更低浓度样品。 实际样品的分析 取大鼠血浆进样,色谱图见图5-1、图5-2。 图5-1大鼠血浆中氢氯噻嗪 图5-2大鼠血浆中尼群地平 精密度与准确度 取大鼠血浆适量,精密配制含有高中低三个浓度的尼群地平和氢氯噻嗪的血浆样品,当天每隔两个小时取 100pL进样,共3次,进行 HPLC分析,考察日内精密度。另选择2天,精密配制含有高中低三个的尼群地平和氢氯噻嗪的血浆样品,每天测一次,考察日间精密度。尼群地平测定精密度见表3,氢氯噻嗪测定精密度如表4所示,由表可知样品日间精密度和日内精密度均小于2.5%,精密度良好,样品测定准确。 表3尼群地平精密度 (n=3) 浓度 日内精密度 日间精密度 (ng/mL) C±SD RSD (%) C±SD RSD (%) 10 9.52±0.18 1.9% 9.20±0.20 2.2% 50 0.9% 51.20±0.51 1.0% 500 496.34±4.96 1.0% 484.36±5.81 1.2% C±SD 52.83±0.48 浓度 日内精密度 日间精密度 (ng/mL) C±SD RSD (%) RSD (%) 20 21.04±0.21 1.0% 20.82±0.27 1.3% 200 198.51±1.59 0.8% 192.11±1.73 0.9% 2000 2032.54±10.16 0.7% 2011.13±20.11 1.0% 回收率 取大鼠血浆适量,精密配制含有高中低三个浓度的尼群地平和氢氯噻嗪的血浆3份,取100 pL 进样,进行 HPLC 分析。尼群地平的回收率见表5,氢氯噻嗪回收率见表6。由表可知尼群地平的回收率较高,重现性好。氢氯噻嗪的回收率在高浓度时稍低,但符合测定要求。分析原因主要是氢氯噻嗪的流出时间与基质流出时间相近, SPE 柱对氢氯噻嗪的保留能力较弱,当大量血浆蛋白存在时,其在 SPE 柱上的保留受基质的干扰,故回收率偏低,对于高浓度样品相对更明显。 表5尼群地平回收率(n=3) 浓度(ng/mL) C± SD 回收率(%) RSD (%) 10 9.52±0.18 92.0% 1.9% 50 52.83±0.48 104.4% 0.9% 500 496.34±4.96 94.5% 1.0% 表6氢氯噻嗪回收率(n=3) 浓度 (ng/mL) C±SD 回收率(%) RSD (%) 20 21.04±0.21 94.9% 1.0% 200 198.51±1.59 84.9% 0.8% 2000 2032.54±10.16 80.2% 0.7% 结论 本研究利用戴安公司UltiMate 3000双三元梯度液相色谱系统建立了同时检测氢氯噻嗪和尼群地平的分析方法。该方法方便,准确,重现性好,适合对血浆样品定量分析。用CAPCELL MF C8固相萃取柱对血浆进行在线前处理,避免手动样品前处理带来的误差,样品基质干扰少,此分析方法可以为进一步的药代动力学-药效学联合模型建立提供支持。 ( 参考文献 ) ( 《中国药典2005版》 第 二 部,氢氯噻嗪 ) ( 《中国药典2005版》 第 二 部,尼群地平 ) DGLC-M-05在线固相萃取-高效液相色谱-紫外检测法测定血浆中的抗真菌药 关键词:在线固相萃取;高效液相色谱;紫外检测;血浆;抗真菌药物 DGLC-M-05 Analysis of Antimycotic Drugs in Biofluids by On-LineSPE-LC with UV detection Key words: on-line SPE; HPLC;UV detection; biofluids; Antimycotic Drugs 引言 以酮康唑为代表的抗真菌药用于抵抗各种真菌疾病。定量测定血浆中此类药物可用于治疗监测。传统测定血浆中此类药物一般要经过沉淀蛋白/离心,液液萃取,过滤以及离线 SPE处理,操作繁琐、费时,样品通量低,费用高昂。 目前,在线 SPE-HPLC技术正好可以克服上诉缺点。 本文描述了 Dionex 公司提供的在线 SPE-HPLC 技术测定血浆中抗真菌药物的方法。 测试条件 仪器: Ultimate DGP 3600系列,包括有带在线脱气单元的双三元梯度泵;配有2500 pL半制备进样组件的自动进样器;带有一个六通阀的柱温箱;紫外检测器。 在线过滤装置: FK7400 (Recipe GmbH, 德国慕尼黑) 分析柱: Acclaim 120C8,3 um, 150x4.6 mm 保护柱: Acclaim 120 C8, 5um, 10×4.3mm 富集柱: LiChrospher ADS RP-4, 25 um, 25×4mm, 截留分子量15000道尔顿 柱温:40℃ 检测波皮:260nm 进样量:100uL 淋洗液组成及流速:见表1; 梯度淋洗条件:见表2. 表1 淋洗液组成及流速 left pump(分析泵) ight pump(富集泵) 流动相A 纯水 流动相B 乙腈 流动相C 0.01 mol/L 醋酸铵 表2 梯度淋洗条件 时间 (min) Left Pump(分析泵) Right Pump (富集泵) 阀 流速 (mL/min) A% B% C% 流速 (mL/min) A% B% C% 0.0 1.2 0 45 55 2 98 2 0 1-2 2.0 0 45 55 2.1 1 1-6 9.0 0 85 15 98 2 0 9.1 10 90 0 1-2 10.0 0 45 55 10 90 0 10.1 2 98 2 0 13 0 45 55 98 2 0 样品前处理方法 血浆样品保存在-20℃冰箱中,分析前在15000 g 下离心10 min。取上清液,待测。 结论 该方法需要两种关键技术: 一是需要采用限制性媒介固相萃取(RAM-SPE)小柱。此种小柱的填料为多孔硅胶或者交联聚合物,具有体积排阻(SEC)和反相作用(RP),能够阻挡血浆中大分子(如蛋白质,核酸,多糖)进入分析柱并同时保留药物成分,从而降低血浆中大分子的干扰并延长分析柱寿命。 二是需要两个梯度泵以及能与之相应的色谱工作站。两个梯度泵中,一个梯度泵用来做on-line SPE; 一个泵用来平衡分析柱并进行分析。色谱工作站要能够满足控制双泵协同作用、 控制阀切换等功能。而戴安公司的双三元高效液相色谱仪以及 Choromeleon 色谱工作站恰恰能满足这个要求。 在线 SPE-HPLC的基本过程如图1:样品首先载入 SPE 柱上进行净化富集(图1左),净化富集完毕后转移到分析柱上(图1中),然后进行分离检测(图1右)。 图1典型的On-line SPE-HPLC过程 本方法可以使血浆中的四种抗菌素:酮康唑,伏立康唑,伊曲康康以及其代谢产物1-羟基伊曲康唑在13min 中内完成分离(图2及图3)。 使用RAM-SPE 固相萃取柱,以每针进样 100uL 体积计,可以重复进样 300次。 1伏犬康唑 1015 ug/L; 2 酮康唑980 ug/L;31-羟基伊曲康唑1254 ug/L; 4 伊曲康唑 1327ug/L 图2空白血浆加标色谱图 1伏立康唑 322.9ug/L; 31-羟基伊曲康唑 24.3ug/L;,4伊曲康唑74.3ug/L 图3服用药物病人血浆色谱图 戴安中国有限公司 香港总部 香港新界葵涌兴芳路223号 新都会广场1座16楼1618-1619室 电话: (852)24283282 传真:(852)24287898 E-mail: dionex@dionex.com.hk 北京代表处 北京市朝阳区安定路33号 化信大厦A座606室 邮编:100029 电话: (010)64436740 (010)64436741 传真::(010)64432350 E-mail: beijing@dionex.com.cn 上海代表处/维修站 上海淮海中路1号 柳林大大2311室 邮编:200021 电话:(021)63735493 (021)63735348 传真::(021)63848294 E-mail: shanghai@dionex.com.cn 应用研究中心 北京市海淀区双清路18号 中科院生态环境中心 邮编:100085 电话: (010)62849182 传真:(010)62849239 E-mail: Dionex App@dionex.com.cn 上海应用中心 上海市张江高科哈雷路 1133办公楼407室 电话:(021)58957001 维修服务中心 北京市朝阳区安定路33号 化信大厦A座606室 邮编:100029 电话: (010)64436740 (010)62936510 传真:(010)62923552 E-mail: service@dionex.com.cn 广州联络处/维修站 广州市天河区天府路237号 华建大厦C座906室 邮编:510630 电话:(0(02200))85613258 传真:(020)85613258 E-mail: penghong@dionex.com.cn 成都联络处/维修站 四川省成都市顺城大街308号 冠城广场8楼F座 邮编:610017 电话::(028)86528208 传真:(028)86528204 E-mail: chengdu@dionex.com.cn 广西联络处/维修站 南宁市民族大道38-2号 泰安大厦金座2102室 邮编:530022 电话:(((0771)5889801 传真:(0771)5889609 E-mail: liugangqian@dionex.com.cn 戴安公司客户服务专专:400-610-0104 400邮箱:400@dionex.com.cn 中文网址: www.dionex.com.cn iso Dionex products are designed,developed,and manufactured under an ISO 9001 Quality90001System. 2011 Dionex Corporation. All trademarks and registeredtrademarks are the property of Dionex Corporation. ·· ··
确定











































还剩93页未读,是否继续阅读?

产品配置单

赛默飞色谱与质谱为您提供《人血浆中苯芴醇检测方案(液相色谱仪)》,该方案主要用于全血/血清/血浆中苯芴醇检测,参考标准--,《人血浆中苯芴醇检测方案(液相色谱仪)》用到的仪器有赛默飞优谱佳UHPLC+高效液相色谱系统
相关方案
更多
该厂商其他方案
更多