挥发性有机物测定,甲醇中苯系物都是7种物质,没有异丙苯,只有二硫化碳种苯系物有8种的标准溶液,大家都买的甲醇还是二硫化碳的呢?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测N,N-二甲基苯胺的浓度可以用二硫化碳做溶剂吗?为什么?
急求苯中二硫化碳标准溶液或者质控样。哪位有相关信息,请告诉我啊,不胜感激!
用二硫化碳来稀释甲醇中的苯,稀释时却发现溶液分层?问了一个同事,说是因为CS2中含有用来液封的水。但是水水可以溶解于甲醇里的啊?另:我在CS2原试剂瓶中也没有看到分层。奇怪?另:加热后,二硫化碳稀释甲醇中的苯溶液又不会分层,静置后又分层?
以前,用二硫化碳萃取法做空气中苯系物的时候,我都是用二硫化碳来稀释浓度比较高的标准品做成标准系列的溶液,做起来挺麻烦,还浪费试剂。因为配好的标准系列很容易挥发,而且二硫化碳中的苯系物混标不好买。 这两天通过在这里查阅了一些做苯系物的文章,我发现了一个不错的方法。就是用甲醇中TVOC的标准品替代二硫化碳中的苯系物混标做标准系列来做标准曲线,分析的时候后,还是用二硫化碳萃取样品后分析。 一、这种方法使的色谱柱必须是毛细管柱。因为填充柱在分析时,甲醇的保留时间苯的保留时间接近,无法分离出苯峰来。而毛细管柱分析时,甲醇和二硫化碳的保留时间都非常早,只是有轻微的拖尾对苯有点影响。 二、甲醇溶液比较稳定。可以用含量高的标准溶液配成100mL的贮备液,作曲线的时候可以取少量的贮备液稀释成标准系列。就算是一次没做好,还可以再重新做。如果是二硫化碳的贮备液就很难保存,或者重新配置了。 三、甲醇的挥发性比二硫化碳低,毒性也比二硫化碳能低点,最咱做化验的来说,安全第一[em31] 不知道,有没有人做过这样的实验?觉得这种方法的准确性如何?可操作性如何? 我准备就这个方法做做实验,大概步骤是——用甲醇中的TVOC标准溶液配成标准系列,做出苯、甲苯、二甲苯的曲线。然后用我手头上的二硫化碳溶剂的苯系物混标来检测一下曲线。 等我做完实验,我会将实验结果传上来分享。不知道谁有这个兴趣能跟我一起做做这个实验? 共同讨论这种方法的优劣。
请教一下各位:2%硫的二硫化碳溶液如何配制,谢谢!
最近做空气中的正丁醇,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,活性碳管采集,2%的异丙醇的二硫化碳溶液解析,最近配的解析液都很浑浊,不知是什么原因,难道是最近温度太低?希望大家帮忙分析一下原因,谢谢!
各位大神,最近滴定分析涉及到重氮偶合反应,做下来总觉得哪里不对劲,而且数据也不太平行,分析下来可能是对硝基苯胺盐酸盐标准溶液出了问题,请问各位大神,配置对硝基苯胺盐酸盐时注意事项有哪些???
我做大气中苯测定时,使用分析纯二硫化碳萃取,在FID毛细管柱中苯的峰面积中夹在未知峰?他和苯保留时间略有变化,我认为是二硫化碳杂质,是二硫化碳出现问题,请老师指导?谢谢
步骤:取1ML甲醛于100ML的浓硫酸中,混匀后作为甲醛-浓硫酸萃取液。取市售的二硫化碳250ML于500ML分液漏斗中,加入20ML的甲醛-浓硫酸萃取液,振荡5分钟后分层(注意及时放气)。经多次萃取到二硫化碳呈无色后,然后用H2O洗至CS2溶液呈中性(用PH试纸测)无水硫酸钠脱水,将溶液倒入试剂瓶中,再加入无水硫酸钠(NA2SO4)垫底约一到二厘米.重蒸馏取46到47度的馏分(火不要开太大)。
职业卫生检测中的正丁醇中用到异丙醇溶液2%的二硫化碳溶液出现浑浊?做出的空白曲线是正常的只有两个溶剂峰,异丙醇加进去就会浑浊,应该不是因为二硫化碳上层的水吧,水是会分层的。
怎异丙醇的二硫化碳溶液2%么配的???请大神指点!1
请问相关同事: 有做苯系物的吗?小弟做二硫化碳中7种苯系物,最近找不到那个标准证书了,也就不知道浓度了。现在只知道批号(332814),请有此溶液相同批号的朋友告诉哈我里边每种物质的浓度。谢谢你们!
GB 11737-89‘二硫化碳用5%的浓硫酸甲醛溶液反复提取,直至硫酸无色为止,用蒸馏水洗二硫化碳至中性,再用无水硫酸钠干燥,重蒸馏,贮于冰箱中’,有做环境空气中苯系物检测的老师可以指导一下二硫化碳的纯化吗
18883-2022中附录C.2中二硫化碳解析苯,曲线所有浓度点都没有问题,采样进行热脱附有峰,但是二硫化碳解析就不出苯峰,二硫化碳解析后溶剂峰没问题,但是苯系物都没有?
各位大师,快帮帮忙啊。做二硫化碳时,二乙胺需要提纯,查了些资料说是要先加入甲苯磺酰氯,乙胺与二乙胺沉淀下来,过滤掉三乙胺;再在沉淀中加入氢氧化钠,乙胺反应掉,二乙胺不反应。但问题是我需要的是二乙胺溶液啊,怎么将二乙胺从沉淀中分离出来呢
各位大师,快帮帮忙啊。做二硫化碳时,二乙胺需要提纯,查了些资料说是要先加入甲苯磺酰氯,乙胺与二乙胺沉淀下来,过滤掉三乙胺;再在沉淀中加入氢氧化钠,乙胺反应掉,二乙胺不反应。但问题是我需要的是二乙胺溶液啊,怎么将二乙胺从沉淀中分离出来呢
新手求助!我利用溴化钾窗片的红外液体池测溶液的红外光谱过程中,使用四氯化碳作为溶剂能够测量,但是使用二硫化碳作为溶剂测量时,溴化钾窗片表面出现溶解(腐蚀)的现象,想请教一下可能是什么原因导致? 我使用的液体池是可拆式液体池,垫片厚度为0.2 mm,二硫化碳为分析纯(试剂上标注的纯度为99.5%)。我在之前查资料时并没有看到二硫化碳不能用溴化钾窗片的,是二硫化碳本身就能溶解溴化钾窗片吗?还是我买的二硫化碳的纯度不够呢?或者各位大神有没有遇见过类似的情况,如何解决的呢? 另外,我在查资料时看到,四氯化碳在800-750 cm-1处有吸收峰,所以一般用二硫化碳互补,如果不能用二硫化碳的话一般选用什么溶剂与其互补呢?
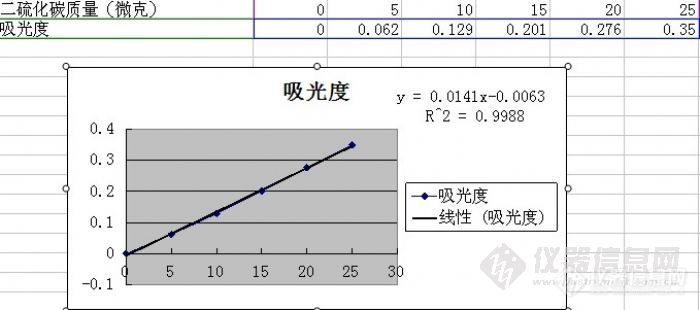
之前,在论坛上发过此贴,是自己去年的实验经历与心得,有几位版友建议参加原创大赛,我有重新整理了一下,与大家分享,欢迎指正。检测依据:GBZ/T 160.33-2004《工作场所空气有毒物质测定 硫化物》8 二硫化碳的二乙胺分光光度法实验用品:http://ng1.17img.cn/bbsfiles/images/2012/10/201210180753_397404_1681389_3.jpg溶剂解析瓶http://ng1.17img.cn/bbsfiles/images/2012/10/201210180754_397407_1681389_3.jpg溶剂解析型活性炭管标准液本实验用的是苯中的二硫化碳,询问了标物中心及标准样品研究所均没有,所以只能自配,一时没有找到高纯的二硫化碳只有分析纯的,当时想先试试,看试一下,看显色过程是否正常,而且标准纯度超过95%,应该影响不大,没想到给自己挖了一个大坑,2011.8.111.标液配制:称0.1564克分析纯二硫化碳,置于事先加入约10毫升苯的50毫升容量瓶中,用苯定容。此溶液浓度为3.128毫克/毫升。2.标准工作液:取此溶液0.80mL用苯定容至50毫升,此溶液浓度为50.05微克/毫升。按标准要求试验不显色。分析原因:标准中写硫酸铜未注明是无水硫酸铜还是五水硫酸铜,可能是铜离子含量低,在溶液中加固体五水硫酸铜,仍不显色。加入分析纯二硫化碳后立即显色,加入上述3.128毫克/毫升的溶液也显色。分析可能是标准溶液配制有问题重新配制后仍不显色。重新配制显色液,仍不显色。2011.8.24最后落实原因:所用分析纯二硫化碳试剂分层,换成环保专用二硫化碳后实验现象正常标准曲线:比色波长435nm 1cm比色皿http://ng1.17img.cn/bbsfiles/images/2012/10/201210181119_397471_1681389_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/10/201210180759_397411_1681389_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/10/201210180801_397414_1681389_3.jpg标准中曲线第一个非零浓度点二硫化碳浓度为5微克,按照GBZ2.1-2007限值要求,限值点按实验操作含量为3微克,感觉不合理,我们想可以有两个途径:第一是减少解析液总体积,标准要求用5毫升苯解析,取0.5毫升检测,我们可以用2毫升解析,取5毫升检测,标准曲线不变,第二是用5毫升解析,取1毫升解析液检测,标准曲线工作液浓度由50微克/毫升变为25毫克/毫升,体积由0.5毫升变为1毫升。首先,我们采用解析液为2毫升苯的方法测定解析效率。2011.8.26二硫化碳解吸率实验配制浓度为10000微克每毫升的二硫化碳标准溶液,分别取3微升、6微升、10微升加入溶剂解析性活性炭吸附管中(将标准溶液自活性炭吸附管进气端注入,同时另一端以30ml/min流量抽气5min后封闭2端静置过夜)。用2ml苯解析后,取0.5毫升分析,同时将相应体积的二硫化碳标液注入2毫升苯中,混匀后,取0.5毫升分析。10微升点:1.前段吸光度0.173 后段吸光度0.007 2. 前段吸光度0.207 后段吸光度0.0066 微升点:1.前段吸光度0.066 后段吸光度0.004 2. 前段吸光度0.108 后段吸光度0.0063微升点:[
用二硫化碳做溶剂,结果二硫化碳峰不出,苯系物的峰都出了,这是怎么回事,这曲线能用吗[img]https://ng1.17img.cn/bbsfiles/images/2019/09/201909161633478638_4360_4000542_3.png[/img]
按HJ 584-2010标准做环境空气中8种苯系物,溶剂是二硫化碳,柱子前面做完顶空后割了10厘米,刚开始走两针,基线比较平,后面用同样浓度走多次,基线飘移越飘越高,这是什么原因啊?[img]https://ng1.17img.cn/bbsfiles/images/2019/03/201903301657004402_4424_3617100_3.png[/img]
11737方法中二硫化碳提取苯、甲苯、二甲苯的检出限能做到多少啊?标准溶液(二硫化碳溶剂)要怎样使用和保存呢?
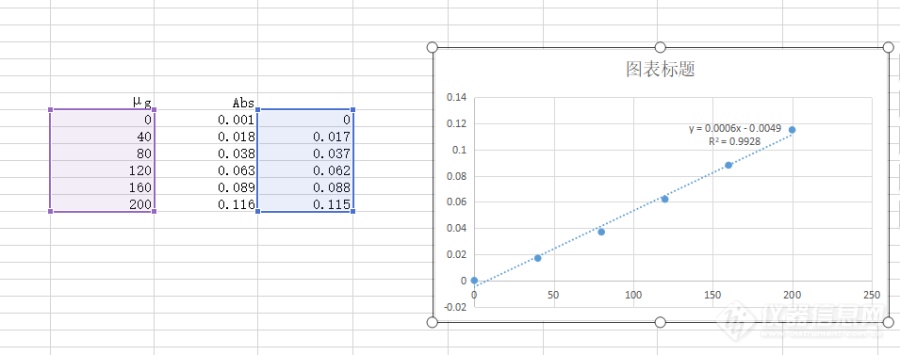
有没有二硫化碳的标准曲线参考一下?我做的曲线吸光度很低,线性也很差,二乙胺和三乙醇胺都是分析纯,二乙胺没有提纯,用的科密欧的无苯级二硫化碳配的标液,配制过程是称取0.0400g二硫化碳用无水乙醇定容到50ml容量瓶配成储备液,再取5ml储备液用无水乙醇定容到50ml容量瓶,算出来标准使用液的浓度是80μg/ml,我试过配成10μg/ml的使用液做曲线但是吸光值很低没有颜色梯度,这一次配成80的浓度勉强能看出点颜色梯度但是线性也不好。。[img=,690,272]https://ng1.17img.cn/bbsfiles/images/2022/01/202201181847008977_5382_3515878_3.png!w690x272.jpg[/img]
在做工作场所空气中有毒物质检测时,一般要用挥发性强的二硫化碳配制标准溶液系列,请问是用普通的移液管多次移液还是用电子移液器分级移液呢。因为母液中的二硫化碳易挥发,而配制标准系列要移液的次数多,用普通的移液管多次移液配制标准系列,总担心浓度偏差,造成线性不好。不知大虾们平常都是怎么处理这种问题的,谢谢!!!
亚砷酸钠溶液和硫化钠混合后加入二硫化碳水层溶液变成橙色了是怎么回事[img]https://ng1.17img.cn/bbsfiles/images/2024/01/202401021029106207_4201_5259927_3.png[/img]
二硫化碳沸点46.25℃,折光率1.631 9,相对密度1.2632。二硫化碳为有毒化合物,能使血液神经组织中毒。具有高度的挥发性和易燃性,因此,使用时应避免与其蒸气接触。对二硫化碳纯度要求不高的实验,在二硫化碳中加入少量无水氯化钙干燥几小时,在水浴55℃~65℃下加热蒸馏、收集。如需要制备较纯的二硫化碳,在试剂级的二硫化碳中加入0.5%高锰酸钾水溶液洗涤三次。除去硫化氢再用汞不断振荡以除去硫。最后用2.5%硫酸汞溶液洗涤,除去所有的硫化氢(洗至没有恶臭为止),再经氯化钙干燥,蒸馏收集。
最近要求做水质苯系物能力验证,做过气中苯系物,想请教各位大神,这个水中苯系物曲线用买的标液来配置,溶剂用甲醇还是二硫化碳?用二硫化碳萃取法,需要用水先进行稀释,再进行萃取,该选哪一种柱子比较好呢?本人小白一枚,希望各位大神回复一下,谢谢!
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]苯。[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]新手。之前做的一直都是二硫化碳中的苯系物,现在来了一个甲醇中的苯盲样,然后做了甲醇中的苯系物标曲,只有6个峰,没有苯的峰,甲醇溶剂峰太大可能把苯的峰给遮蔽了。我现在准备用二硫化碳萃取甲醇中苯,请教各位老师这种方法可行么?值的大小影响大么?
做环境空气中苯系物的时候,用二硫化碳解吸,苯的峰分成两半了,有时候不分,有时候分,标液(二硫化碳中苯系物)走出来也是这样,有没有大佬分析下是什么情况[img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108100926585253_264_5343586_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108100926588522_8346_5343586_3.png[/img]
HJ 584-2010环境空气苯系物的测定中规定用二硫化碳做解吸液,而现在标样所中所出售的苯系物的标准溶液都是甲醇基体的,买回来后用二硫化碳稀释可以吗?对结果有什么影响







 我要推广仪器
我要推广仪器
 下载APP
下载APP