¥3万 - 15万
中航时代
暂无样本
DSC差示扫描量热仪-DSC差示扫描量热分析仪
--
中国大陆

看了差示扫描量热仪(DSC/DTA)的用户又看了
1.1 全新的炉体结构,更好的解析度和分辨率以及基线稳定性;
1.2 仪器下位机数据实时传输,界面友好,操作简便。
DSC | DSC-214 | DSC-204 | DSC-404 | DSC-214H | DSC-404H |
DSC量程 | 0~±600mW | ||||
温度范围 | RT~600℃ | -40℃~-600℃ | -150℃~-600℃ | RT~600℃(带降温扫描) | -150℃~600℃(带降温扫描) |
升温速率 | 0.1~100℃/min | ||||
温度精确度 | ±0.01℃ | ||||
温度准确度 | 0.001℃ | ||||
温度波动 | ±0.01℃ | ||||
温度重复性 | ±0.1℃ | ||||
DSC精确度 | 0.001mW | ||||
DSC解析度 | 0.001mW | ||||
工作电源 | AC220V/50Hz或定制 | ||||
控温方式 | 升温、恒温、降温(全程序自动控制) | ||||
程序控制 | 可实现六段升温恒温控制,特殊参数可定制 | ||||
曲线扫描 | 升温扫描、降温扫描、曲线扫描 | ||||
气氛控制 | 两路自动切换(仪器自动切换) | ||||
气体流量 | 0-300mL/min(可定制其它量程) | ||||
气体压力 | ≤0.55MPa | ||||
显示方式 | 24bit色7寸LCD触摸屏显示 | ||||
数据接口 | 标准USB接口 | ||||
参数标准 | 配有标准物质(锡),用户可自行矫正温度和热焓 | ||||
仪器热电偶 | 三组热电偶,一组测试样品温度,一组测试内部环境温度,一组炉体过热自检传感器 | ||||
软 件 | 带有温度多点校正功能 | ||||
设备尺寸 | 500*500*300(mm)(长宽高) | ||||
备注 | 所有技术指标可根据用户需求调整 | ||||
_副本.jpg")
_副本.jpg")
_副本.jpg")

热力学基础
3.1 引言
热分析法是在一定气氛和程序控制温度下, 测量物质的物理性质随温度或时间变化的一系列技术的总称。通过实验测量得到的物理性质随温度或者时间变化的曲线, 通常称为热分析曲线。由于热分析曲线是物质在不同温度下的性质的实时反映,因此通过热分析曲线可以得到与热力学相关的一些重要信息。
热力学是一门古老的学科,是从宏观角度研究物质的热运动性质及其规律的学科。热力学属于物理学的分支, 其与统计物理学分别构成了热学理论的宏观和微观两个方面。热力学主要是从能量转化的观点来研究物质的热性质, 揭示了能量从一种形式转换为另一种形式时所遵循的宏观规律,总结了物质的宏观现象而得到的热学理论。在实际应用中,其在解决不同的反应过程中反应物的温度和热量方面发挥着重要的作用。在希腊语中, 热力学(thermodynamics)术语中“thermos”具有热量的含义,而“dunamis”则具有能量的含义。事实上,温度的概念也是在热力学中定义的,
因此,使用热分析技术或量热技术的相关人员应该对热力学知识有较为全面和深入的了解。本章将简要地介绍一些与热分析相关的热力学背景知识。
3.1.1 热力学状态
通常用热力学相关理论来研究宏观体系,热力学定律也只适用于足够大的宏观体系。因此,从理论上来说,在研究热力学时不需要考虑体系中物质的微观结构。
经验表明,当一个宏观的体系长时间处于孤立状态时, 体系的状态将不会随时间而发生改变,通常称这个状态为平衡态(equilibrium state)。理论上,可以通过一系列的宏观物理量如体积、压强和温度等描述该平衡态,而平衡态与非平衡态(non-equilibrium state)是有明显的区别的。在热力学中,没有必要讨论与时间相关的物理量,通常用热力学中的过程(process)来描述体系从一个(平衡)状态到另一个(平衡)状态的变化。
3.1.2热力学状态函数
通常将描述体系的宏观可测量性质状态的热力学性质变量分为广度性质(又称广延性质或容量性质)(extensive property)和强度性质(intensive propcrty)两大类。
广度性质与强度性质都属于体系的状态性质,即状态函数,二者均需满足状态函数的一切性质,即:
(1)当热力学状态一定时,体系的热力学状态函数的数值也一定, 即状态函数是体系状态的单值函数。状态函数的数值仅与体系所处的状态有关,而与其过去的历史无关。
(2)当体系的状态发生变化时,状态函数的变化值只决定于体系的初始状态和最终状态,而与其由初始状态到最终状态所经历的具体路径和方式无关。
(3)状态函数的微小改变量可以用全微分方程的形式来表示。
(4)体系恢复原状,状态函数也将恢复至初始状态的数值。
(5)体系的各个状态函数之间相互制约,其中的几个状态函数一旦确定,则其余的状态函数也可以随之确定。
广度性质是指体系中和体系大小或体系中的物质的量成比例改变的物理性质, 其具有加和性,即整体的性质是组成整体的各部分的性质之和。也就是说,如果将一个体系分成两个子体系,那么两个子体系的状态与原来的体系的状态相同,两个子体系如体积和质量数量的值之和等于原始体系数量的值。常见的与体系中存在的物质的量成正比的热力学变量主要有体积V、质量m、物质的量n、热力学能U、焓H、熵S、吉布斯自由能G、亥姆蛋兹自由能A等。
在数学上,广度性质是物质的量的一次齐次函数。对于一个由i种物质构成的均匀体系而言,其中每种物质的物质的量是n1,n2,…,ni,体系的状态可以用(T,p,n1,…,ni)描述。当物质的量n1,n2,…,ni改变α倍时,物理量F相应地改变α倍,即满足数学关系式
时,则称F为广度性质。
另一方面,强度性质则与体系的数量无关。强度性质是指体系中不随体系的大小或体系中物质的量的多少而改变的物理性质,强度性质是尺度不变的物理量。强度性质是指数值上与体系中物质的量无关的性质,即其不具有“部分加和性质”,其数值仅取决于体系自身的特性。在平衡体系中任一强度性质的数值与体系中任一部分的该强度性质的数值都相等。例如将一金属块分成两部分后,每一块金属的温度不会发生改变,与整块金属的温度相等。需要强调指出,强度性质虽然不具有“部分加和性”,但是其可以具有“组分加和性”,即整个体系的强度性质是体系中各个组分的该强度性质的总和,例如, 体系的总压强可以由各个组分的分压加和得出。
在数学上,强度性质是物质的量的零次齐函数。对于由i种物质构成的一个均匀体系,每种物质的量是n1,n2,…, ni,体系的状态是由(T,p,n1,…, ni)描述的。当物质的量n1,n2,…, ni改变α倍时,物理量F不发生变化,即满足数学关系式
时,则称F为强度性质。
主要通过以下原则来区分强度性质与广度性质:
(1)通过将一个体系分成几个部分,性质具有加和性的为广度性质。
(2)如果各部分的性质都相同,则为强度性质。也就是说,如果将体系划分为两个子体系,则对描述强度性质的状态和数值均不会产生影响。常见的强度性质主要包括压强、密度和化学组成等。
另外,还可以用以下方式对比广度性质与强度性质的差异。测量广度性质时必须考虑整个体系,而强度性质则取决于体系中一个确切的位置。在一个体系中,不同位置的强度性质不一定是一样的。例如,对于一个由水和一些冰块组成的体系而言,不同位置的冰或水的密度显然是不相同的。
体系中的两个广度性质的状态函数的比值为强度性质的状态函数, 例如摩尔体积是由体积和物质的量这两个具有广度性质的状态函数相比得出的,其为强度性质的状态函数。
与热力学(此处特指“经典热力学”或“平衡热力学”)密切相关的是统计热力学(statistical thermodynamics)、不可逆热力学(irreversible thermodynamics)和动力学(kinctics)。在统计热力学中.主要研究宏观热力学函数与物质的分子结构之间的联系,而不可逆热力学则主要用来描述处于非平衡状态的体系, 因此在非平衡热力学中时间起着十分重要的作用。动力学则通常用来描述与时间相关的过程(参见本书第4章)。
3.2 热力学体系与温度的概念
3.2.1 热力学体系
热力学体系(thermodynamic system)又称热力学系统,是指用于热力学研究的有限的宏观区域,是热力学的研究对象。体系的外部空间被称为该体系的环境(surroundings)。所研究的体系与环境之间通过一个边界(boundary)分隔开,这个边界既可以是真实存在的,也可以是虚拟的,但必须将所研究的体系限制在一个有限空间内。为了便于理解,可以将以上所述的边界理解为一堵“墙”(这堵“墙”可以是真实的,也可以是虚拟的)。体系与其环境之间通过边界进行物质、功、热或其他形式能量的传递。体系、界面与环境的关系如图3.1所示。
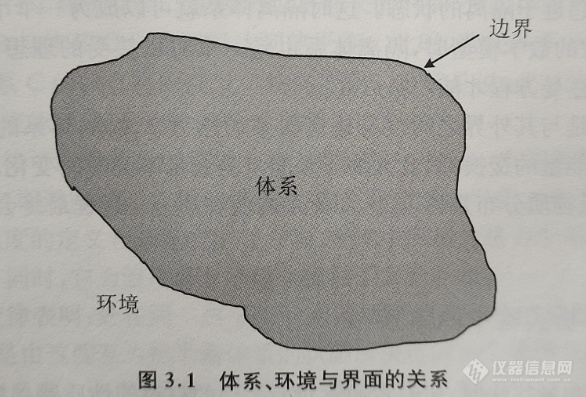
按照热力学体系的边界所允许传递的量的不同,可以将体系分为以下三类。
3.2.1.1 开放体系
开放体系(open system)也称敞开体系, 体系与环境之间通过界面进行物质和热量的交换。在开放体系中,通过其边界的一些部分与外界进行物质传递,而边界的其他部分则可能只允许进行能量传递而不允许物质传递,同时还需要考虑体系内部各部分之间的能量传递过程。对于一个热力学过程而言,体系的边界和环境的性质都十分重要,因为它们决定了某一过程是否能够顺利进行。通常,开放体系的边界允许物质进行传递。而当考虑到体系内能的变化时,体系与环境之间的物质传递则需要进行热能和机械能之外形式的能量的传递。
对于热重实验而言,当实验过程中发生质量变化时, 体系与环境之间进行了物质的传递过程,此时一般将其视作开放体系。有时为了处理方便,也会将产生的气体与残余的固态或者液态物质一起看做一个体系。这样一来,体系与环境之间不存在物质的传递,可以当做封闭体系处理。
3.2.1.2 封闭体系
封闭体系(closed system)是一种可以与其环境传递能量(热量或功)但不能传递物质的热力学体系。例如,通常可以将温室看做一个封闭体系,其可以与其环境交换能量,但不能与其环境传递物质。体系边界的性质决定着能量的传递形式,即一个体系与外界传递的是热量或机械能或二者可以同时进行传递。实际上,对于相当多的量热仪所研究的对象可以视作封闭体系。
在通常情况下,如果没有特别的说明,所指的体系即为一个封闭体系。
当体系的边界绝热,即其不允许热量传递时,则称该体系为绝热体系。而当体系的边界为刚体,即其不允许机械功传递时,则称该体系为力学孤立体系。
3.2.1.3 隔离体系
隔离体系(isolated system)也称孤立体系,是彻底孤立于其环境的热力学体系。也就是说,体系与周围环境之间完全隔离开来,其与周围环境之间既没有物质的交换,也不存在热量的交换。而在现实中,由于体系的内部与外界之间多少会存在像万有引力这样的相互作用的力,因此彻底孤立于其环境的热力学体系并不存在。然而,现实中的热力学体系可以在一定的时间内处于超近于隔离的状态。这时隔离体系就可以成为一个十分有用的模型。另外,在建立某些现象的数学模型时,隔离体系也是一个可以接受的理想化模型。例如,只有在隔离体系中玻尔兹曼方程才能严格成立。
由于隔离体系是与其外界之间没有任何联系的热力学体系, 体系的边界不允许其与环境之间进行物质和能量的交换, 因此其物质和能量的总量不随时间变化。另外, 隔离体系的内部压强、温度以及物质分布是均匀的, 而体系的所有均一化过程最终会使体系达到热力学平衡。
3.2.2热分析实验中的体系
在使用热分析和量热法时,研究者通常对所关注的材料的性质感兴趣。实验时, 用来研究的物质的量是确定的,在实验过程中所使用的样品可以是纯的化学物质,也可以是由两种或多种化学物质组成的混合物。
众所周知,一种纯物质可以以固态、液态和气态的不同的聚集状态形式存在,有时也称这些聚集的状态为“相”(phase)。更严格意义上的“相”的定义为:相是由具有相同强度性质的体系所组成的。如果一个体系由一个单个相组成,则称这个体系为均相体系(homogeneous system)。如果一个体系由两个或多个相所组成,则该体系就被称为非均相体系(heterogeneous system)。在非均相体系中,两个不同的相之间通过界面(interface)彼此分开。通常一个体系只有一个气(或蒸汽)相,但可能会存在几个不同的液相和固相。例如,对于一个由水和油的混合物组成的体系而言,其由两个不同的液相组成。一个相由少量溶解的油和水组成,而另一个相则由少量溶解的水和油组成。即使是由单一物质组成的固态体系,也有可能会存在多个固态相。例如,当一定量的氯化铯加热到温度为743K时,可以观察到其从一个固态相到另一个固态相的相转变(这两个固相之间的区别主要表现在晶体结构的不同上)。在转变温度(743K)时,两个相可以同时共存,呈现出非均相平衡状态。
3.2.3 温度的概念
温度(temperature)是表示物体冷热程度的物理量,微观上,温度是组成物质的分子的热运动的剧烈程度的反映。从分子运动论观点看,温度是物体分子运动平均动能的标志。温度是大量分子热运动的集体表现,具有统计意义。宏观上,温度是根据某个可观察现象(例如水银柱的热胀冷缩)按照几种温度标度之一所测得的冷热程度的反映。实际上,只能通过物体随温度变化的某些特性来间接测量温度,而用来量度物体温度数值的标尺称为温度标准(temperature scale),简称温标。温标规定了温度的读数起点(零点)和测量温度的基本单位,温度的国际单位为热力学温标(K)。目前国际上应用较多的其他温标有华氏温标、摄氏温标和国际实用温标。
对于热力学体系而言,如果两个不同的体系都处于内部的平衡状态,此时若将这两个体系进行热量交换,则它们的热力学状态将会发生变化。在经历了一段时间之后,这两个体系的状态不再发生改变,此时这两个体系彼此间达到了热平衡状态。现在我们考虑三个体系,分别用字母A、B和C表示。假设体系A与体系B之间进行了热量交换,与此同时体系B和体系C之间也进行了热量交换。在经历了足够长的时间之后,体系A与体系B之间将会达到热平衡,体系B与体系C也将同时达到热平衡。此时, 若使体系 A与体系C进行热接触,则体系A与体系C的状态将不会发生任何变化。也就是说,此时体系A与体系C也达到了热平衡。这个规律通常被称为“热力学第零定律”。
热力学第零定律可以概括为如果两个热力学体系中的每一个体系都与第三个热力学体系处于热平衡状态(即温度相同),则它们彼此也必定处于热平衡。热力学第零定律的重要性在于它给出了温度的定义和温度的测量方法,定律中所指的热力学系统是指由大量分子、原子组成的体系。同时,它为温度概念的建立提供了实验基础。
热力学第零定律表明:处在同一热平衡状态的所有的热力学体系都具有一个共同的宏观特征,这一特征是由这些互为热平衡体系的状态所决定的一个数值相等的状态函数,这个状态函数被定义为温度。注意:温度相等是热平衡的必要条件。
因此,当所有的体系与一个明确定义的参考体系进行接触并且彼此之间达到热平衡时,可以用温度来描述所有的体系处于热平衡状态。
3.2.4 温度的测量及温标
3.2.4.1早期的温度测量
为了测量温度,需要一个定义准确的温标,以及一个可测量的与温度相关的量。最早的温标利用液体的体积依赖于温度的变化以及纯物质具有恒定的熔点和沸点的性质, 最早的温度计为液体-玻璃温度计(图3.2),这种温度计将少量的液体放置在一个玻璃槽里,并且其可以膨胀到一个较细的毛细玻璃管中。在标定温度时,需将玻璃槽放进一个热交换良好且具有确定的温度值的体系(通常用一种纯物质的熔点和沸点作为参考温度)中,然后标定玻璃管中液体上升的高度。用同样的方法可以标定第二个确定的温度值,然后在这两个已经确定的温度刻度值之间除以温度差来得到单位温度的刻度值。
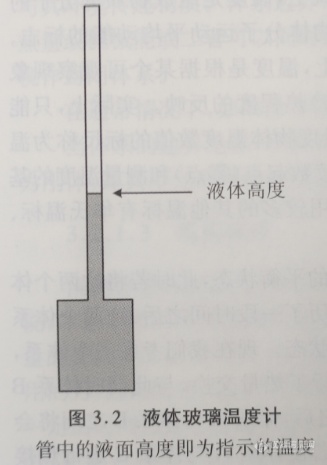
大约在1700年,丹麦科学家Ole Romer用这种装置进行了实验,他使用酒精作为膨胀的介质。德国科学家D.G. Fahrenheit在1708年参观了Ole Romer的实验室之后,发明了世界上第一个温度计。他开始用的材料是酒精,之后在1714年用水银替代了酒精。
3.2.4.2 温标的定义
温度标准,简称温标,是以量化的数值加上温度的单位来表示温度的方法。它也是温度计进行刻度的根据。理论上,通过物理方法使环境中产生两个不同的温度,对其进行测量并赋予不同的数值,即可以确定温标。例如:摄氏温标定义水的熔点和沸点分别为0℃和100℃(详见摄氏温标)。
温标的定义包括以下三个要素,即:
(1)选定测温物质及其测温属性,此属性通常用数值表示,即某种物质的测温参量(如铂的电阻、热电偶的温差电动势等):其中,测温物质为测量温度所用的物质,主要有:
①电阻温度计:根据金属丝的电阻随温度变化来标记温度。热电阻是中低温区最常用的一种温度检测器。它的主要特点是测量精度高、性能稳定。其中铂热电阻的测量精确度是最高的,它不仅广泛应用于工业测温,而且其可以被制成标准的基准仪。
热电阻的测温原理是基于金属导体的电限值随温度的增加而增加的这一特性来进行温度测量的。热电阻大都由纯金属材料制成,目前应用最多的是铂和铜。此外,现在已开始采用镍、锰和铑等材料制造热电阻。热电阻测温系统一般由热电阻、连接导线和显示仪表等组成。
②热电偶温度计:利用两种金属导体组成的热电偶的电动势随温度变化的原理来测量温度,热电偶是工业上最常用的温度检测元件之一。其优点是:
a.测温精度高。热电偶直接与被测对象接触,不受中间介质的影响。
b. 测温范围广。常用的热电偶从-196到1600℃均可连续测量,某些特殊热电偶最低可测到-269℃(如金铁镍铬),最高可达2800C(如钨-铼)。
c.构造简单,使用方便。热电偶通常由两种不同的金属丝组成, 而且其不受大小和形状的限制,外有保护套管,用起来非常方便。
热电偶测温的基本原理为:将两种不同材料的导体或半导体A和B焊接起来,构成一个闭合回路。当导体A和B的两个接触点之间存在温度差时,两者之间便产生电动势,因而在回路中形成一个电流,这种现象称为热电效应。热电偶就是利用这一效应来工作的。
常用的热电偶可分为标准热电偶和非标准热电偶两大类。
标准热电偶是指国家标准规定了其热电势与温度的关系、允许误差, 并有统一的标准分度表的热电偶,有与其配套的显示仪表可供选用.
非标准化热电偶在使用范围或数量级上均不及标准化热电偶,一般也没有统一的分度表,主要用于某些特殊场合的测量。
我国从1988年1月1日起,热电偶和热电阻全部按IEC国际标准生产,并指定S、B、E、K、R、J、T七种标准化热电偶为我国统一设计型热电偶。
由于热电偶的材料一般都比较贵重(特别是采用贵金属时),而测温点到仪表的距离都很远。为了节省热电偶材料,降低成本,通常采用补偿导线把热电偶的冷端(自由端)延伸到温度比较稳定的控制室内,连接到仪表端子上。热电偶补偿导线的作用只起延伸热电极,使热电偶的冷端移动到控制室的仪表端子上,它本身并不能消除冷端温度变化对测温的影响,不起补偿作用。因此,还需采用其他修正方法来补偿冷端温度t0≠0℃时对测温的影响。在使用热电偶补偿导线时必须注意型号相配,极性不能接错,补偿导线与热电偶连接端的温度不能超过100℃。
③光学式高温计:光学式高温计一般指不与高温物体接触,而是通过对它的辐射热量来测量温度,根据热辐射公式可以推算出该物体温度的一类仪器,因其可不必与被测物体接触,故可应用于测量极高温度、运动热源温度以及不能接触或对人体和仪器有害的热源温度。
一般来说,光学式高温计主要有以下两种结构形式:
在光学式高温计中,灯丝加热到标定的热度,与高热物体的颜色相比较,调整灯丝热度,使其颜色与高热物体相一致,灯丝的温度即为高热物体的温度。
在辐射式高温计中,将来自物体的辐射集中在温差电堆(thermopile)上,根据其所截获的热辐射量产生相应的电压,再将电压换算为该物体的温度。
(2)确定测温参量与温度之间的关系(在尚未确立任何温标之前,这种关系只是在一定经验的基础上做出的假定关系)。测温参量为测温物质的某一随温度变化的属性, 即确定的测温参量与温度的函数关系。在最简单的情况下,测温参量与温度呈正比, 例如在理想气体温标中,当压强趋近于0时,气体温度计的气体体积与其热力学温度成正比;测温参量也可以与温度呈线性、对数、指数等复杂的函数关系。
(3)确定标准温度点并规定其数值,亦即具有确定的标度方法。在1954年的国际计量大会上选取水的三相点作为标准温度点,并规定此状态下的温度为273.16K(0.01℃),记为Ttr。
任何一种温标都包含有以上三个方面的确定内容(热力学温标不涉及测温质,属于例外情况),改变其中的任何一个方面即成为另一种温标。常用的温标主要有华氏温标、摄氏温标、开氏温标、理想气体温标、热力学温标等,其中华氏温标、摄氏温标和开氏温标属于经验温标,理想气体温标属于半理论性温标,热力学温标则属于理论性温标。
3.2.4.3 华氏温标
华氏温标由Fahrenheit于1714年建立。他最初规定氯化铵与冰的混合物为0°F;人的体温为100°F。后来规定在标准状态下纯水与冰的混合物为32°F;水的沸点为212°F。两个标准点之间均匀划为180等份,每份为1°F。
3.2.4.4 摄氏温标
第二个被广泛使用的温标为摄氏温标,由Celsius于1742年建立。最初,他将水的冰点定为100℃,水的沸点定为0℃, 如今我们使用的摄氏温标则是他的学生S.Martin 在1749年所创立的。1960年国际计量大会对摄氏温标做了新的定义,规定它由热力学温标导出。摄氏温度(符号t)的定义为
3.2.4.5 兰氏温标
兰氏温标又称华氏绝对温标,是美国工程界使用的一种温标,单位用°R表示。该温标规定在一大气压下水的冰点为491.69°R,沸点为671.69°R。开氏温标以水的三相点为273.16K,兰氏温标以273.16K作为491.688°R。它们都是从绝对零度起算,因此兰氏温标又称华氏绝对温标。
华氏温度tF与兰氏温度TR之间的换算关系如下:
3.2.4.6 开氏温标(也称开尔文温标)
开氏温标由开尔文于1848年建立。1954年国际计量大会规定水的三相点的温度为273.16K。这个数值的规定有以下的历史原因:
(1)开尔文温标的每一度的温度间隔与早已建立并广为使用的摄氏标度法每一度的间隔相等。
(2)按理想气体温标,通过实验并外推得出理想气体的热膨胀率为1/273.15。由此确定-273.15℃为绝对温度的零度,而冰点的绝对温度为273.15K。
(3)将标准温度点由水的冰点改为水的三相点(相差0.01℃)时,按理想气体温标确定的水的三相点的温度就确定为273.16K。
3.2.4.7 理想气体温标(即绝对温标)
一个非常重要的温标是理想气体温标(ideal gas temperaturescale),亦即绝对温标(absolute temperature scale)。这个温标是基于无限稀释的理想气体的性质而创立的。玻意耳(Boyle)在1660年的研究成果表明,对于一定量的稀释气体而言,在一定的温度下,它的压强和体积的乘积是恒定的。盖吕萨克(Gay-Lussac,1778~1850)发现:对于恒定体积下的稀薄气体体系而言,其温度和压强呈线性关系;在恒定的压强下,其体积和温度呈线性关系。可以将这些观察结果与玻意耳定律相结合,推导出以下的玻意耳-盖吕萨克定律(The Law of Boyle-Gay-Lussac)
式中,p为气体的压强;V为气体的体积;t为摄氏温度;C和α为常数。常数C与气体的量有关,α=1/273.15。
等式(3.3)表明,当t=-273.15℃时,p和V的乘积等于0。因此,不存在比-273.15℃更低的温度,因为那时压强或体积的值将会变为负数。等式(3.3)还表明,我们可以定义一个绝对温标(这种温标的温度单位是开尔文, 通常用K 表示), 该温标的零点(即绝对零度)为-273.15℃。绝对零度是一个设定的值, 并且仅有一个可以选择的固定的参考值(而其他的温标至少有两个固定的参考值可以选择)。为了使水的凝固点和沸点之间存在 100 K的温度差,水的凝固点温度被定义为T=273.15K。当建立绝对温标(也称为理想气体温标)之后,可以用下式来描述玻意耳-盖吕萨克定律:
式(3.7)即为理想气体定律(the ideal gas law)。式中,p为气体的压强,单位为Pa;V为气体的体积,单位为m3;T为绝对温度,单位为K;n为物质的量, 单位为mol;R为气体常数(R=8.31451J·K-1·mol-1)。
与经验温标相比,理想气体温标优点在于其与任何气体的任何特定性质无关。不论用何种气体,当外推到压强为零时,由它们所确定的温度值都一样。但是,理想气体温标毕竞还要依赖于气体的共性,其对极低温度(氦气在低于1.01×105Pa的蒸汽压下的沸点1K以下)和高温(1000℃以上)并不适用。另外,理想气体温标在具体操作上也不够便捷。
3.2.4.8 热力学温标
19世纪下半叶,汤姆孙(W. Thomson)(开尔文勋爵)提出了热力学温标(thermodynamic temperature scale)的概念。这种温标已被证明与理想气体温标是等效的。
在热力学温标中,热量Q起着测温参量的作用,然而比值Q1/Q2(Q1为可逆热机从高温热源吸收的热量;Q2为可逆热机向低温热源放出的热量)并不依赖于任何物质的特性。因此,热力学温标与测温物质无关。
当然,任何一种温标都必须是某种测量依据与某种标度法的结合。通常,任何一种温标都可以用于不同的测温物质的某种测温参量, 如水银摄氏温度计、酒精摄氏温度计;任何一种测温参量也都可以采用不同的标度法,例如理想气体开尔文温标、理想气体摄氏温标。但是以热量Q为测温参量的热力学温标,其标度法仅为开氏标度法,所依据的是热力学第二定律,这是它与其他温标的根本不同之处。
表3.1中给出了以上介绍的几种温标之间的换算关系。
表3.1 几种温标之间的换算关系
温标 单位 | 开尔文T(K) | 摄氏度t(℃) | 华氏度θ(°F) | 兰氏度t(°R) |
K | 1 | T-273.15 | (T-273.15)×1.8+32 | 1.8T |
℃ | t+273.15 | 1 | 1.8t+32 | (t+237.15) ×1.8 |
°F | (θ-32)/1.8+273.15 | (θ-32)/1.8 | 1 | θ+459.67 |
°R | t/1.8 | t/1.8-273.15 | t-459.67 | 1 |
3.2.4.9 1990年的国际温标
对于以符号T和单位K表示的热力学温标而言,其为一个基本的物理量。然而,在实际上这种温标却很难得到广泛应用。同样地,由于实际气体与理想气体的性质存在着许多的差异,与热力学温标等同的绝对温标或者理想气体温标也很难在实际中得到广泛应用。因此,在实际上通常使用由国际度量衡委员会(the International Committee of Weights and Measures)经过对一系列点的检测和插值分析而确定的统一温标。最近一次确定的统一温标是1990年的国际温标(International Temperature Scale of 1990, ITS-90)。ITS-90温标的温度值是与热力学温度非常近似的值。
概括起来,ITS-90温标主要包括以下几个方面的内容:
(1)以热力学温标为基本温标。
(2)热力学温度以符号T表示,单位为开尔文,简称为开,符号为K。
(3)1K的大小定义为水的三相点热力学温度的1/273.16。
(4)由热力学温度导出摄氏温度(符号为t)的规定,其定义为t(℃)=T(K)-273.15。摄氏温度的单位称摄氏度,符号为℃,其大小与开尔文相同。
(5)划分了四个温度范围,指定了以下各温度范围的基准温度计:
①0.65~5.0K。在此温度范围,基准温度计为3He、4He蒸气压温度计。
②3.0~24.5561K(氖的三相点),在此温度范围,基准温度计为3He、4He定容气体温度计。
③13.8033(平衡氢的三相点)~1234.93K(银的凝固点)。在此温度段,基准温度计为铂电阻温度计。
④1234.94K以上,基准温度计为光学高温计。
另外,ITS-90温标还定义了17个标准温度点,如表3.2所示。
表 3.2 ITS-90 温标定义的17个标准温度点
物质状态 | 温度 | |
T90(K) | t90(℃) | |
氮在1标准大气压下的沸点 | 3~5 | -270.15~-268.15 |
平衡氢的三相点 | 13.8033 | -259.3467 |
平衡氢在25/26标准大气压下的沸点 | ≈17 | ≈-256.15 |
平衡氢在1标准大气压下的沸点 | ≈20.3 | ≈252.85 |
氖三相点 | 24.5561 | -248.5939 |
氧三相点 | 54.3584 | -218.7916 |
氩三相点 | 83.8058 | -189.3442 |
汞三相点 | 234.3156 | -38.8344 |
水三相点 | 273.16 | 0.01 |
镓熔点 | 302.9146 | 29.7646 |
铟凝固点 | 429.7485 | 156.5985 |
锡凝固点 | 505.078 | 231.928 |
锌凝固点 | 692.677 | 419.527 |
铝凝固点 | 933.473 | 660.323 |
银凝固点 | 1234.93 | 961.78 |
金凝固点 | 1337.33 | 1064.18 |
铜凝固点 | 1357.77 | 1084.62 |
3.2.5 微观尺度的温度
在之前的内容中所讨论的温度是一个宏观的量。当然,在宏观的热力学温度性质和微观的分子性质之间有一个桥梁来进行联系。虽然在3.1节中已经指出体系的微观结构对于热力学处理来说不是必要的,但通过微观结构的信息仍然有利于我们理解相关的理论。
理想气体模型是对于非常稀薄气体所做的一个微观模型假设。在这个模型中,分了之间不存在相互作用力。假设分子之间和分子与器壁之间的碰撞为弹性碰撞,每个分子以一定的速度做随机运动,由此可以计算得到分子的动力学能量。可以通过一个函数来描述分子运动速度的分布情况,据此可以计算出气体的动力学总能量和由于分子与器壁碰撞所引起的器壁的压强变化。在得到这些信息后,可以进一步推导出压强、体积和动力学总能量之间的关系。在与理想气体状态方程(等式(3.7))进行比较之后可以得出这样的结论:在宏观的温度下,理想气体体系的总动力学能量与温度有关。
对于其他体系而言,温度与体系中分子的热运动有关。
3.3 热力学定律
3.3.1 热力学第一定律
3.3.1.1热力学第一定律的本质
热力学第一定律的本质是能量守恒与转换定律, 其为自然界的基本规律之一。自然界中的一切物质都具有能量,能量不可能被创造,也不可能被消灭;但能量可以从一种形态转变为另一种形态,且在能量的转化过程中能量的总量保持不变。我们知道,运动是物质的属性,能量是物质运动的度量。分子运动学说闹明了热能是组成物质的分子、原子等微粒的杂乱运动即热运动的能量。既然热能和其他形态的能量都是物质的运动,那么热能和其他形态的能量可以相互转换,并在转化时能量守恒完全是理所当然的。热力学第一定律是能量守恒与转换定律在热现象中的应用, 它确定了热力学过程中热力学体系与外界进行能量交换时,各种形式的能量在数量上的守恒关系。
热力学第一定律是人类在实践中累积的经验总结,它不能用数学或其他理论来证明,但第一类永动机迄今仍未造成以及由第一定律所得出的一切推论都与实际经验相符合等事实,可以充分说明它的正确性。
3.3.1.2 热力学第一定律的基本内容
通过之前的内容我们已经了解到,可以通过对体系加热和/或对体系做功来改变体系的状态。人们在过去就已经充分地认识到,在某些情况下体系可以只通过被加热或只通过对其做功来实现相同的状态变化。例如,在初始温度下一定量液体的状态变化就属于这种情况。可以通过加热使这个体系的温度稍微升高。通过放置一个搅拌器在该隔离体系的液体中,并对搅拌器做功使其温度发生变化。也可以对体系加热,使其达到相同的最终温度。焦耳在1840年通过这个实验测出了热功当量(mechanical equivalent of heat)的关系,即增加的热量与做的功成比例。正是这种实验结果,证明了热量和功可以用同一单位表示。
经验表明,对于每个(封闭的)体系而言,当其达到最终状态时,无论通过什么样的路径,添加到体系的热量的总和以及对体系所做的功的总量是恒定的,这就是著名的热力学第一定律(The First Law of Thermodynamics)。
3.3.1.3热力学第一定律的数学表达式
在数学上,可以用热力学函数来描述体系所处的状态。当体系的状态发生改变时, 则热力学函数值的变化量应等于对体系所施加的热量和对体系所做的功的总和。由于热量和功是能量的转移形式,因此这个热力学函数通常被称为体系的内能(internal energy,通常用符号U表示)。当体系从状态A变化到状态B时,则可以用下式的形式来表示热力学第一定律:
式中,Q为环境对体系传递的热量;W为环境对体系所做的功。
由于Q和W受状态变化过程中所经历的路径的影响,因此其不属于热力学状态函数的范畴。
如果除做功、传热外,还有因物质从外界进入体系而带入的能量Z,则式(3.8)可以表示为
从等式(3.8)和等式(3.9)可以看出,只有能量差是可以通过测量某一过程的热量和功来确定的。尽管我们可以描述体系某个状态的能量,但是必须首先确定一个特定的能量作为已知参考点。在化学热力学中,通常定义采用的能量参考点如下:将化学元素在温度为298.15K和压强为1bar(或者1标准大气压)的条件下的能量设定为0。
由于能量函数依赖于体系的状态,因此内能(以及所有其他依赖于体系状态的热力学函数)称为状态函数。与此相反,热量和功很明显与体系的状态无关,它们仅仅是与过程相关的量。与能量差函数相反,热量和功也与其所经历的一个过程有关,它们不仅仅依赖于体系的初始和最终状态,也依赖于过程的路径。
在通常的条件下,观察体系状态的无穷小的变化是很方便的。例如,内能的无穷小的变化可以由内能的微分得到,用dU表示,对于在过程中所增加的热量和对体系所做的功而言,依赖于所选择的路径(即热量和功不是体系的状态函数),把它们表示成微分形式的物理量是不合适的,因此没有无穷小的热和功的微分表达形式。
对于无穷小的体系状态变化而言,可以用下式的形式来描述热力学第一定律:
如果除了体积功之外没有其他的做功形式,则对体系做的无穷小量的功而言,其可以表示成下式的形式:
3.3.1.4 恒容过程的热力学第一定律的数学表达式
对于恒容过程而言,由等式(3.11)可知,在该过程中并没有对体系做功。这意味着从等式(3.8)或者等式(3.11)可以得到,体系内能的变化量等于体系热量的变化量, 其关系式可表示如下:
以上形式的这个结论非常重要,因为对于恒定体积下的所有过程来说,可以通过量热仪来测量体系与周围环境交换的热量q,热量与体系内能的变化直接相关,且内能与变化的路径无关。
在此基础上,Berthelot研制出了可以用来测量“燃烧热”的氧弹,氧弹是体积恒定的钢制容器。在实验时,将已知质量的样品和过量的氧气放置于其中,点燃样品,通过测量燃烧过程中的温度差的变化即可得到燃烧热。使用这种方法,还可以同时测量得到物质中各组成元素的燃烧热。通过化学物质的组成,可以计算出物质和各组成元素之间的能量差。
3.3.1.5 恒压过程的热力学第一定律的数学表达式
对于恒容过程而言,体系的热量变化等于内能的变化这个结论十分有用。然而,在实际上有许多过程发生在恒压(如大气压强)条件下。很明显,对于这样的过程而言,体系所做的功并不等于0,因此交换的热量并不等于内能的变化。在这种情况下, 需要重新定义一个与内能密切相关的新的物理量。这个物理量称为焓(enthalpy, 通常用 H 表示), 焓被定义为内能与压强与体积的乘积之和,用下式表示:
显然,焓和内能一样,也是一个状态函数。焓具有能量的量纲,一定质量的物质按定压可逆过程由一种状态变为另一种状态,焓的增量便等于在此过程中吸入的热量。
当体系的状态发生了无穷小的变化时,无穷小的焓的变化可以表示为下式的形式:
结合热力学第一定律(等式(3.10))和体积功的表达式(3.11),可得
对于恒压过程而言,体系与环境之间所交换的热量等于体系的焓变,即
这意味着,在恒压过程中,可以将由测量所得到的热量与一个状态函数(即焓)直接相关联起来。因此,焓的变化与路径无关。基于这个原因,在过去一段时间曾经错误地将焓称为热含量(heat content)或者含热量。
焓与内能一样,其绝对值无法确定。在统计热力学中虽然可由分子配分函数计算出焓值,但这样求得的也是焓的相对值,因为物质内部的运动形式不可穷尽,不可能计算出所有运动形式的配分函数。
焓是与内能有关的物理量,反应在一定条件下是吸热还是放热由生成物和反应物的焓值差即焓变(ΔH)决定。在化学反应过程中所释放或吸收的能量都可用热量(或换成相应的热量)来表示,通常称为反应热,又称“焓变”,可由量热法测量得到。焓是一个状态量,焓变是一个过程量。
一般来说,在发生热效应时,有如下关系:
(1)若体系吸热,则焓值升高;若体系放热,则焓值降低。
(2)对于均匀体系的简单状态变化而言, 由于吸热时体系的温度升高, 因此高温物质的焓要高于低温物质的焓。
(3)对于相变化,固体变为液体,固体变为气体和液体变为气体都要吸收热量,因此在同一温度下处于聚集状态的同种物质的焓值不相等,且有H(g)>H(1)>H(s)。
(4)对于等温下的化学反应而言,若反应吸热,则产物的焓高于反应物的焓;若反应放热,则产物的焓应低于反应物的焓。
3.3.1.6 热容
在工程和材料科学中,热容(heat capacity)是一个重要的物理量,常用符号C来表示。体系的热容可以定义为:在不发生相变化和化学变化的前提下,向体系中加入(无穷小的)热量将会引起的(无穷小的)温度变化,其可以用(无穷小的)热量与(无穷小的)温度升高的比值形式来表示:
添加到体系一定的热量,使其完成一个特定的状态,该过程中的变化所需的热量取决于变化的途径。因此,热容也取决于变化的途径。
体系与环境所交换的热量的多少应与物质的种类、状态、物质的量和交换的方式有关。因此,体系的热容值受上述各因素的影响。另外,温度变化范围也将影响热容值,即使温度变化范围相同,体系所处的初始、结束状态不同,体系与环境所交换的热值也不相同。因此,由某一温度变化范围内测得的热交换值计算出的热容值,只能是一个平均值,称为平均热容。热容的单位为J·K-1,热容是体系的广度性质。1mol物质的热容称为摩尔热容,以Cm表示,单位为J·K-1·mol-1,C=n·Cm;单位质量物质的热容称为比热容。
对于恒容过程而言,可以通过联立等式(3.12)和等式(3.17)得到恒容热容(通常用 Cv表示),如下式所示:
这意味着,在恒定体积下的热容是内能对温度的偏导数。
对于恒压过程而言,可以由联立等式(3.16)和等式(3.17)得到恒压热容(通常用Cp表示),如下式的形式:
这意味着,恒定压强下的热容是焓对温度的偏导数。
如果热容已知,则可以通过下式来计算体系在加热(或降温)过程中体系内能的变化或焓变:
由以上可见,热容是温度的函数,热容值随温度的变化范围不同而不同。当状态变化的范围较小时,热容实际上可视为常数。当温度趋于绝对零度时,各种物质的热容都趋近于零。
许多研究者用实验方法精确地测定了各种物质在各个温度下的热容值,求得了表示热容与温度关系的经验表达式。通常采用的经验公式有下列两种形式:
以上两式中,a、b、c、c′均为经验常数,随物质的不同及温度变化范围的不同面异。各物质的热容经验公式中的常数值可以查询有关的参考书及手册得到。
3.3.2 热力学第二定律
3.3.2.1 热力学第二定律的一般性表述
热力学第二定律(The Second Law of Thermodynamics)是由法国工程师萨迪·卡诺(S.Carnot)在1824年提出的,时间在热力学第一定律之前。为了叙述方便,通常把热力学第一定律放在第二定律的前面来进行介绍。
在卡诺提出了卡诺定理后,德国人克劳修斯和英国人开尔文在热力学第一定律建立以后重新审查了卡诺定理,意识到卡诺定埋必须依据一个新的定理,即热力学第二定律,他们分别于1850年和1851年提出了克劳修斯表述和开尔文表述,这两种表述在理念上是等价的。
热力学第二定律可以表述为:热不可能从低温物体传到高温物体而不产生其他影响(克劳修斯表述),或不可能从单一热源取热使之完全转换为有用的功而不产生其他影响(开尔文表述),或孤立系统的熵永远不会自动减少,熵在可逆过程中不变(熵增加原理)。
3.3.2.2 热力学第二定律的数学表述
体系与周围环境之间所交换的热量不仅取决于体系所处的初始状态和最终状态, 也与其所经历的路径有关。然而,可以证明,对于可逆过程而言,由无穷小的热量交换所引起的热量的减少并不依赖于所经历的过程,可以用无穷小的热交换量与热力学温度的比值来表示这种形式的热量减少,如下式所示:
以上的表达式实际上为热力学第二定律的进一步描述,其虽然在这里作为一个假设的形式被提出, 但其已被所有的后续的实验所证实。在表达式中使用的温度是绝对热力学温度。这种温标与体系的任何特殊性质无关,可以证明热力学温标与理想气体温标是等效的。
对于可逆过程而言,体系减少的热量与所经历的过程路径无关,我们因此可以定义一个新的状态函数。通常称这个状态函数为熵(entropy,通常用符合S表示,由克劳修斯于1865 年首次提出),可以用以下形式的表达式来表示一个状态变化:
或者对于无穷小的变化而言,用以下表达式表示微小的熵变:
对于体系而言,在经历了一个熵变的过程之后,如果这个过程是可逆的,只能通过测量(变化的)热量并应用等式(3.25)来确定熵的变化。如果该过程是不可逆转的,由于熵是状态函数,因此可以假设一个初始状态和最终状态不同的可逆路径,并使用等式(3.25)来计算熵变。
熵是热力学中用来表征物质状态的热力学函数,其物理意义是体系混乱程度的度量。在孤立体系中,体系与环境之间不存在能量交换,体系总是自发地向混乱度增大的方向变化,最终使整个系统的熵值增大,即熵增原理。还可以从一个自发进行的过程来考察该过程:热量Q由高温(T1)物体传至低温(T2)物体,高温物体的熵减少dS1=δQ/T1,低温物体的熵增加dS2=δQ/T2。当把两个物体合起来当成一个体系时,熵的变化是dS=dS2-dS1>0,即熵是增加的。
在分子层面上,对于体系中的分子而言,其平动速度、转动速度、振动能量等等都在不断地发生着改变,体系在一定时刻的分子状态由该时刻每个分子所有这些量的值决定。对于一个由宏观的能量、体积等物理量决定的热力学状态而言,其包含了体系在过去的时间中大量的分子“构象”(configurations)状态。如此看来,在热力学物理量熵和体系中指定能量和体积下可能存在的分子构象的数量W之间似乎存在着一定的联系。根据玻尔兹曼(Boltzmann)在1872年提出的理论,可以用以下的关系式来表示这种关系:
式中,k为玻尔兹曼常数,k=R/NA=1.380658×10-3J·K-1,NA为阿伏伽德罗常数(Avogadro's constant),NA=6.0221367×1023mol-1,指1mol物质中分子的数量。
这意味着一个具有大量的可能状态数的分子体系比分子状态数更少的体系具有更大的熵值(即体系更加有序)。由此可见,水的熵大于冰的熵(当熔点T=273.15K时)。
3.3.2.3热力学第二定律的解释
热力学第二定律是热力学的基本定律之一,其表明(在自然状态下)热量永远都只能由热处传到冷处。它是关于在有限空间和时间内,一切和热运动有关的物理、化学过程具有不可逆性的经验总结。
在Clausius对热力学第二定律的定义中,指出了在自然条件下热量只能从高温物体向低温物体转移,而不能由低温物体自动向高温物体转移,也就是说在自然条件下,这个转变过程是不可逆的。如果使热传递方向倒转过来,只有靠消耗功来实现。
在开尔文对热力学第二定律的定义中,自然界中任何形式的能都会很容易地变成热, 而反过来热却不能在不产生其他影响的条件下完全变成其他形式的能,从而说明了这种转变在自然条件下也是不可逆的。热机可以连续不断地将热变为机械功,在该过程中一定伴随有热量的损失。第二定律和第一定律不同,第一定律否定了创造能量和消灭能量的可能性,第二定律则阐明了过程进行的方向性,否定了以特殊方式利用能量的可能性。
从分子运动论的观点看,做功是大量分子的有规则运动,而热运动则是大量分子的无规则运动。显然无规则运动要变为有规则运动的概率极小,而有规则的运动变成无规则运动的概率大。一个不受外界影响的孤立系统,其内部自发的过程总是由概率小的状态向概率大的状态进行,由此可见热不可能自发地变成功。
热力学第二定律仅适用于由很大数目分子所构成的体系及有限范围内的宏观过程。而不适用于少量的微观体系,也不能把它推广到无限的宇宙。
热力学第二定律指出在自然界中任何的过程都不可能自动地复原,要使体系从最终状态回到最初状态必须借助外界的作用。由此可见,在热力学体系中所进行的不可逆过程的初态和终态之间存在着重大的差异,这种差异决定了过程的方向, 通常用状态函数熵来描述这个差异,从理论上可以进一步证明:①可逆绝热过程Sf=Si;②不可逆绝热过程Sf>Si。式中,Sf和Si分别为体系的最终状态和最初状态的熵值。
也就是说,在孤立体系内,对可逆过程而言,体系的熵总保持不变;对不可逆过程而言,体系的熵总是增加的,通常称这个规律为熵增加原理,这也是热力学第二定律的又一种表述形式。熵的增加表示体系从概率小的状态向概率大的状态演变,也就是从比较有规则、有秩序的状态向更无规则、更无秩序的状态转变。熵体现了体系的统计性质。
另外,在有限的宏观体系中热力学第二定律应满足如下条件:①该体系是线性的;②该体系全部是各向同性的。
3.3.3 热力学第三定律
和其他热力学定律一样,热力学第三定律是被一个假想的实验所证实的。在本部分内容中将不对产生这些结论的实验结果做进一步的讨论。
能斯特(Nernst)指出,任何物质在发生物理或者化学变化时的熵变在温度非常接近于绝对零度时都等于0,即
这种表述形式也被称为能斯特热定理(Nernst heat theorem)。普朗克(Planck)简化了能斯特的表述,他指出,当温度接近绝对零度时, 过程的熵变和每种凝聚态物质的实际熵(the actual entropy)也均等于零。在以上的表述中, 将混合物明确排除在外。而在下面的表述中则没有必要使混合物排除在外:对于每个平衡的体系, 它的熵在绝对零度下都为0(For each system in equlibrium, the entropy equals zero when the temperature approaches the absolute zero)。这个表述被称为热力学第三定律(The Third Law of Thermodynamics):
关于热力学第三定律的一个重要推论是:化学物质(包括元素和化合物)的绝对熵(absolute entropy)(即与参考状态无关)的值是可以被确定的。在实验上可以通过量热法测量物质在从绝对零度到指定温度的整个范围的热容和这个温度范围内发生的相变潜热,这样可以确定一种物质的绝对熵值。根据等式(3.25)和等式(3.26),可以按照如下的方式来计算绝对熵值:
在上式中,假设在温度范围为从0到Θ的过程中仅存在一个相变过程(在T=Ttr时,由Ⅰ相转变为Ⅱ相)。
在统计物理学上,热力学第三定律反映了微观运动的量子化。在实际意义上,第三定律并不像第一、二定律那样明确地告诫人们放弃制造第一种永动机和第二种永动机的意图,而是鼓励人们想方设法尽可能地接近绝对零度。现代科学可以使用绝热去磁的方法达到5×10-10K,但永远达不到0K。
根据热力学第三定律,基态的状态数目只有一个。也就是说,第三定律决定了自然界中基态无简并。
3.3.4 热力学第零定律
在本章3.2.3小节中,大致介绍了热力学第零定律。热力学第零定律(The Zero Law of Thermodynamics)又称热平衡定律,是热力学的四条基本定律之一,是一个关于互相接触的物体在热平衡时的描述,为温度提供理论基础。最常用的定律表述为:“若两个热力学体系均与第三个体系处于热平衡状态,则此两个体系也必然彼此间处于热平衡状态。”
热力学第零定律通常用作体系进行温度测量的基本依据,其重要性在于其给出了温度的定义和温度的测量方法。该定律还有以下的一些表述形式:
(1)可以通过使两个体系相接触,并观察这两个体系的性质是否发生变化来判断这两个体系是否已经达到热平衡;
(2)当外界条件不发生变化时,已经达成热平衡状态的体系,其内部的温度是均匀分布的,并具有确定不变的温度值;
(3)一切彼此平衡的体系具有相同的温度,因此可以通过另一个与之平衡的体系的温度来表示一个体系的温度,也可以通过第三个体系的温度来表示。
热力学第零定律是在不考虑引力场作用的情况下得出的,物质(特别是气体物质)在引力场中会自发产生一定的温度梯度。如果两个封闭容器分别装有氢气和氧气, 由于它们的分子量不同,它们在引力场中的温度梯度也不相同。如果在最低处它们之间可交换热量,温度达到相同,但由于两种气体的温度梯度不同,则在高处温度就不相同,也即不平衡。因此第零定律不适用于引力场存在的情形。
综上所述,可得到以下结论:
(1)根据热力学第零定律,可以用来确定温度状态函数;
(2)根据热力学第一定律,可以用来确定内能和焓状态函数;
(3)根据热力学第二定律,可以用来确定熵状态函数。
3.4热力学自由能
3.4.1 简介
热力学自由能(thermodynamic free energy)是指一个热力学体系的能量中可以用来对外做功的部分,是热力学态函数。通常情况下,可以用自由能作为一个热力学过程能否自发进行的判据。
对特定条件不同的热力学过程而言,热力学自由能有不同的表达形式。最常见的有吉布斯自由能G和亥姆霍兹自由能A(或F)。一般来说,等温等容过程用亥姆霍兹自由能A=U-T·S作为自发性判据,而对于等温等压过程则用吉布斯自由能G=H-T·S作为判据,式中H为焓。两者之间存在着G=A+p·V的关系。
3.4.2 吉布斯自由能和亥姆霍兹自由能的引入
对于可逆过程而言,可以用等式(3.24)来表示体系与环境之间的热交换。在引入嫡的概念之后,则等式(3.24)可以用如下形式的等式表示:
用式(3.31)可以表示可逆过程的热交换,用等式(3.11)可以表示可逆过程的体积功,则该可逆过程的热力学第一定律表达式可写成:
在3.5节中将引入状态函数——焓。通过将等式(3.31)代入等式(3.15)中,可以得到以下形式的关于可逆过程(无限小的进行状态下)的焓变关系式:
由于熵是状态函数,温度也是描述状态的一个物理量,则与温度T和熵S的乘积(即T·S)有关的量也是一个状态函数。这意味着如果分别将内能和焓减去乘积T·S之后,则可以得到两个新的状态函数。这两个状态函数分别被称为Helmholtz白由能(Helmholtz energy,用A表示,有时也称作Helmholtz能)和Gibbs白由能(Gibbs energy,用G表示,有时也称作Gibbs能)。
根据这些定义,可以用下式表示无限微小的Helmholtz自由能和Gibbs自由能的变化过程,如下式所示:
3.4.3 热力学基本关系式
对于封闭的均匀热力学体系而言,可以使用温度T、内能U、熵S、焓H、Gibbs自由能G、Helmholtz自由能A、压强p和体积V这八个基本的热力学函数来描述,据其可以从不同的侧面或不同的过程揭示宏观物质所处的体系的宏观性质。通过这些热力学函数的关系式,可以将可以通过实验测量的变量与不可以通过实验测量的变量关联起来。
根据状态函数的全微分方程式的交叉微分的特性,可以推导得出以下形式的等式:
可以证明,若下列一个关系式中的一个表达式已知,则可以求得体系所处状态的所有热力学物理量:
(1)内能与熵、体积的关系式;
(2)焓与熵、压强的关系式;
(3)Helmholtz自由能与温度、体积的关系式;
(4)Gibbs自由能与温度压强的关系式。
因此,通常称这些关系式为热力学基本方程(或者特征方程)。
如果将交叉微分特性应用到内能、焓、Helmholtz自由能和Gibbs自由能的全微分方程式中,则可以分别得到如下形式的等式:
以上这些等式非常重要, 特别是最后的两个等式尤为重要。因为这些等式表明了熵与体积、压强的关系(这在实验中难以测得)和压强或体积与温度的关系是等效的。
3.5 热力学平衡条件
在没有外界影响的条件下,如果某个体系中各部分的宏观性质(如体系的化学成分、各物质的量、温度、压力、体积、密度等)在长时间内不发生任何变化,则称该体系处于热力学平衡状态。从统计热力学的角度来看,体系的宏观性质是相应的微观量的统计平均值。当体系处于热力学平衡状态时,体系内的每个分子仍处于不停的运动状态,体系的微观状态也在不断地发生着变化,只是分子微观运动的某些统计平均值不随时间而改变。因此,热力学平衡是一种动态的平衡状态。
一个热力学体系必须同时达到下述几方面的平衡,才能处于热力学平衡状态。
(1)热平衡
如果体系内部不存在隔热壁,则体系内各部分的温度相等。如果没有隔绝外界的影响,在体系与环境之间不存在隔热壁的条件下,当体系达到热平衡时, 则休系与环境的温度也相等。
(2)力学平衡
如果不存在刚性壁,则体系内各部分之间不存在不平衡的力。如果忽略重力场的影响,则达到力学平衡时体系内部各部分的压强应该相等。如果体系和环境之间不存在刚性壁,则平衡时体系和环境之间也不存在不平衡的力,体系和环境的边界将不随时间而移动。
(3)相平衡
如果体系处于一个非均相状态,则平衡时体系中的各相之间可以长时间地共存,各相的组成和数量都不随时间而改变。
(4)化学平衡
如果体系内各物质之间可以发生化学反应,则达到平衡时体系的化学组成及各物质的数量将不随时间而改变。
在下面的内容中,我们将讨论不同体系的热力学平衡。
3.5.1 封闭体系
封闭体系与外部环境之间只有能量交换,而无物质交换。若要实现一个体系的平衡状态, 通常需要假设一个处于完全隔离状态的体系。假设一个体系发生了一个从状态 A到状态B的不可逆自发过程,由于该体系处于完全隔离状态,则体系和环境之间并没有功和热量的交换,可以用下式表示:
假设该体系发生了一个从状态B到状态A的可逆变化。在此可逆过程中,可以用以下的等式表示体系与环境交换热量以及环境对体系做的功:
对于整个变化过程而言,由于体系的初始状态和最终状态相同,因此其能量的变化为零。根据热力学第一定律(等式(3.8)),可以得到以下形式的关系式:
根据热力学第二定律的一般形式的表达式,体系在吸收一定的热量后并不可能全部转化为功(参见3.4.1小节)。在整个过程中,可以用下式的形式来表示热量由体系转移到环境的过程:
根据热力学第二定律,体系从状态B到状态A可逆过程的熵变可以用下式表示:
在一个完全隔离体系的不可逆过程中,熵一定会增加,存在以下的关系式:
在等式(3.51)中,下标U和V表示在不可逆过程中内能和体积保持不变,这意味着体系在此过程中是完全隔离的。
由于在隔离体系的自发过程中熵增加,并且总会朝着平衡的方向进行,因此可以认为平衡状态对应于最大的熵值。
由于以上这种平衡是在隔离体系中达到的,因此在实际上很难实现这种平衡条件。因此,对于封闭体系而言,我们需要找到一个更切合实际的平衡条件。通常假设一个(巨大的)隔离体系,该体系由一个小的非平衡体系和一个巨大的类似“蓄水池”的稳定的平衡体系共两个子体系组成。于是,隔离体系的总的熵变由小的非平衡体系的熵变和无限大的平衡体系的熵变组成。因此,根据等式(3.51)可以得到整个体系的(无限小的)熵变,其结果为正值,如下式所示:
式中,下标s、1和t分别用来表示小体系、大的稳定平衡体系和整个体系的参数。假设小的非平衡体系的温度Ts与大的稳定平衡体系的温度T1相等,即Ts=T1,并且在整个过程中温度保持不变。由于大的稳定平衡体系始终处于平衡状态,因此该稳定体系的熵变可以用改写后的等式(3.32)来表示,如下式所示;
由于整个体系是完全隔离的,因此整个体系的体积保持不变,总的内能也保持不变,如下式所示:
在以下两种不同的情形下:
(1)在小体系的不可逆过程中,体系的体积保持不变,即dVs=0。
根据等式(3.54),大的稳定平衡体系的体积也将保持不变,即dVr=0。将此等式与等式(3.53)和等式(3.54)联立,并代入等式(3.52)中,可以得到以下的关系式:
由于我们假设了小体系的温度和体积保持不变,因此等式(3.56)可以改写成以下形式的关系式:
引入Helmholtz自由能,由于A=U-T·S,于是等式(3.57)可以变形为
这表明,对于一个等温等体积条件下的非隔离体系中的不可逆过程而言,在此状态变化过程中,其Helmholtz自由能降低。当达到平衡状态时,可以将平衡状态下的Helmholtz自由能视为能量最低的状态。
(2)在小体系的不可逆过程中,其压强和大的稳定平衡体系相同并且为常数,即ps=pr=常数。
将此等式与等式(3.53)、等式(3.54)和等式(3.55)一起代入等式(3.52)中,可以得到以下形式的关系式:
即
由于我们假设了小体系的温度和压强为恒定值,则等式(3.59)可以改写为下式的形式:
在引入Gibbs自由能之后,由于G=H-T·S=U-T·S+p·V,于是等式(3.61)可以变形为
这表明,对于一个等温等压下的非隔离体系中的不可逆过程而言,在此状态变化过程中,其Gibbs自由能降低。当达到平衡状态时,可以将平衡状态下的Gibbs自由能看做能量最低的状态。
3.5.2 开放体系
在以上的内容中,我们讨论了封闭体系(即和环境没有物质交换的体系的热力学平衡条件)。通常称与环境之间存在着物质交换的体系为开放体系,在下面的内容中将讨论开放体系。与封闭体系相比,开放体系通常与环境之间既存在着能量交换,也存在着物质交换。在开放体系的热力学表达式中,通常需要引入一个与组成相关的物理量。
3.5.2.1 多组分均相开放体系
假设一个体系由多个化学成分(在一个均匀相中)组成,并且该体系为均相体系。显然,这个体系的状态与体系中每种化学组分的含量有关系。体系中每个组分的量可以用物质的量(n)来表示,单位为摩尔(mol)。如果每个组分都有确定的数量,则第i个组分的量可以用ni表示。
在多组分均相开放体系中,需要用体系中各个组分的物质的量来表示体系的热力学状态函数表达式。对于一个开放体系而言, 内能除了与熵和体积有关之外, 还与体系中存在的每个组分的量有关。从数学的角度上看,内能的全微分方程式可以改写为下式的形式:
通常称内能对组分i的物质的量ni的偏微分表达式为化学势,也称为热力学势(thermodynamic potentials),用μi表示:
因此,对于一个开放体系而言,等式(3.32)可以改写为下式的形式:
类似地,在开放体系中,可以将焓、Helmholtz自由能和Gibbs自由能改写成下列形式的等式:
根据等式(3.68),化学势可表示为下式的形式:
在等温等压条件下,通常称热力学函数对物质的量的偏导数为偏摩尔量(partial molar quantity)。因此,也可以称化学势为偏摩尔吉布斯自由能。根据全微分方程的交叉微分特性,可以用以下形式的等式来表示化学势与温度和压强的关系:
在以上的等式(3.70)和等式(3.71)中,si和vi分别是i组分的偏摩尔熵和偏摩尔体积。这些偏摩尔量是体系的压强、温度和组分的函数关系式。
当体系中的组分保持不变时,根据以上这些等式,可以用以下形式的等式来表示全微分形式的化学势与温度和压强的关系:
对于在等温条件下的理想气体而言,假设其只由一种组分组成,气体从常压p=p0=1bar被压缩到p=pf。在此过程中,由等式(3.71)与理想气体状态方程式可以计算得到气体化学势的变化Δμ(T),用下式表示:
通常设标准压强p=p0,p0是单位压强(通常用大气压atm表示,p0=1 bar=105Pa或者p0=1atm=101325Pa),用上标“0”表示(有时也用“θ”表示)。通常称标准压强条件下的热力学性质为标准热力学性质,于是可以定义标准化学势为
联立等式(3.73)和等式(3.74),可以得到以下等式:
由于p0经常表示一个大气压,则等式(3.75)通常可以表示为以下形式的等式:
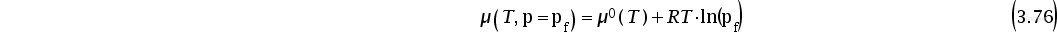 结合Gibbs自由能的定义,根据等式(3.35)及等式(3.69),可以得到化学势与偏摩尔焓(以hi表示)和偏摩尔熵(以si表示)的关系,用下式表示:
结合Gibbs自由能的定义,根据等式(3.35)及等式(3.69),可以得到化学势与偏摩尔焓(以hi表示)和偏摩尔熵(以si表示)的关系,用下式表示:
3.5.2.2 多相热力学平衡体系
1.单组分纯物质体系
随着温度和压强的变化,体系中的单组分物质可以以多种相态的形式存在。对于一种纯物质而言,其通常存在固态、液态和气态三种相态形式。下面以一种物质的熔融过程(即由固态到液态的转变过程)的相变为例来介绍单组分纯物质体系的热力学平衡。该过程可以看做一种物质的固态含量(dnsol)减少,同时液态的含量(dnliq)增加。如果分别用μ*sol和μ*liq (纯物质组分用上标“*”表示)来表示固态相(简称固相)和液态相(简称液相)的化学势,那么根据等式(3.68)可以得到在该过程中Gibbs自由能的无限微小的变化,用下式表示:
由于物质的总量在相变的过程中保持不变,则有如下的关系式:
联立等式(3.78)和等式(3.79),可以得到以下等式:
由于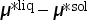 与温度和压强有关(因为化学势与温度和压强有关),因此随着温度和压强的变化将会出现以下三种不同的情形:
与温度和压强有关(因为化学势与温度和压强有关),因此随着温度和压强的变化将会出现以下三种不同的情形:
(1) 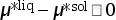 。
。
此时,液相的化学势小于固相的化学势。因此,体系从一定量的固相到液相的转变会引起Gibbs自由能的降低。根据等式(3.67),此过程不可逆且为自发过程。
(2) 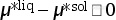 。
。
此时,液相的化学势大于固相的化学势。因此,体系从固相到液相的转变会引起Gibbs自由能的升高。根据等式(3.67),可以判断此过程为自发反应(前提是这种转变不受动力学的限制),Gibbs自由能降低。
(3) 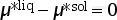 。
。
此时,液相化学势等于固相化学势,体系中一定量的固相到液相的转变(或液固转变)不会改变Gibbs自由能。此时,液相和固相可以共存,即固相和液相之间达到了平衡。
对于一种纯物质而言,最稳定的相态是化学势最低的状态。
由于化学势是压强和温度的函数,因此可以很方便地在压强-温度图中表示纯物质的不同相稳定存在的范围。在通常情况下,将用来表示不同状态下同一物质不同的稳定相态的图称为相图(phase diagram),也称相态图、相平衡状态图。相图是用来表示相平衡系统的组成与一些参数(如温度、压力)之间关系的一种图。相图在物理化学、矿物学和材料科学中具有很重要的地位。广义的相图是在给定条件下体系中各相之间建立平衡后热力学变量强度变量的轨迹的集合表达形式,相图表达的是平衡态,严格来说是相平衡图。由相图不能说明平衡过程中的动力学,不能判断体系可能出现的亚稳相。
单组分纯物质的相图称为一元相图(unary phase diagram)。在此类相图中,将不同的稳定相分开的曲线称为两相平衡曲线(two-phase equilibrium curves), 通常用该曲线表示两相平衡的条件。
“稳定相”(stable phase)是指某一相相对于其他相是稳定的。由于从其他相到稳定相之间的转变使Gibbs自由能降低,因此该过程为一个自发过程。然而,对于存在动力学限制的过程而言,上述这种转变可能不会自发地进行。因此,在相图中会出现某一相虽然在稳定区域之外但仍然可以表现为稳定相态的现象。
例如,如果降低水的温度,在0℃时可得到水-冰两相平衡的曲线。在更低的温度下,冰比水更稳定。在实际上,水可能会在低于零下几度的状态下仍然以液态水的形式存在,以上的这种过冷状态也可以称为亚稳态相。
2.两相平衡(equilibrium between two phases)
假设在压力为pe、温度为Te时的一种纯物质的两个相α相和β相的性质和数量均不随时间变化时,α相和β相彼此之间达到了相平衡,此时的状态称为相平衡态,用相平衡点(pe,Te)表示。此时从宏观上看,没有物质由一相向另一相的净迁移。但从微观上看,不同相间分子转移并未停止,只是两个方向的迁移速率相同。
当达到了相平衡状态时,在此温度和压强下共存的两相的化学势相同,可用下式的形式表示:
假设温度和压强发生了无限微小的变化,可以看做这两相之间此时仍然维持在平衡状态。这可以理解为,温度和压强的改变所对应的状态在两相平衡线上移动,即点(pe+dp,Te+dT)仍然在两相的平衡线上。由于温度和压强发生了改变,因此化学势也随之发生变化,可以通过等式(3.72)来计算化学势的这种变化。如果体系在新的状态下重新达到了平衡,则化学势会再次相等,这说明α相的化学势的变化量与β相的相同,可以表示为下式的形式:
对等式(3,82)进行重排,可以得到下式;
式中,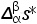 和
和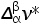 分别为摩尔熵变和摩尔体积变化。由此可以得出,在两相平衡曲线中的压强和温度的变化满足以下关系式:
分别为摩尔熵变和摩尔体积变化。由此可以得出,在两相平衡曲线中的压强和温度的变化满足以下关系式:
等式(3.84)即为Clapeyron方程,
对于α相和β相而言,将等式(3.82)代入等式(3.84)中,可得
联立等式(3.84)和等式(3.85),可以得到另外一种形式的Clapeyron方程式:
由于从α相到β相的转变焓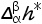 可以通过量热仪器方便地测得,因此以上这种表达形式的Clapeyron方程式更为常用。
可以通过量热仪器方便地测得,因此以上这种表达形式的Clapeyron方程式更为常用。
对于凝聚态(通常为液相和固相)与蒸气相之间的平衡而言,可以通过一些假设来对Clapeyron方程式进行简化。一般情况下,物质在蒸气状态下的摩尔体积远大于处于凝聚相的同一物质的摩尔体积。因此,摩尔体积的变化可以近似看成是蒸气的摩尔体积变化(即凝聚相的摩尔体积变化量可以忽略不计):
如果假设蒸气相具有类似理想气体的性质,则可以根据理想气体方程得出摩尔体积,如下式所示:
将等式(3.87)和等式(3.88)两式同时代入Clapeyron方程式(即等式(3.86)),可得
以上等式也可以变形为
以上这两个非常有用的方程是等价的,通常称之为克劳修斯-克拉佩龙方程式(Clausius-Clapeyron Equation)。该方程表明, 由相平衡曲线(用平衡条件下的压强的自然对数与温度的倒数表示)的斜率可以得到负数形式的汽化或升华焓变对气体常数的比值。虽然汽化焓变(对应于液相与蒸汽相的平衡)和升华焓变(对应于固相与气相的平衡)与温度有关,但是其对温度的导数值变得很小。因此在温度变化范围不是很大的情况下, 可以将升华熵变与汽化熵变看做一个常数。因此,等式(3.90)经积分后可以得到以下的Clausius-Clapeyron 方程的第三种表达形式:
在等式(3.91)中,点(p1,T1)和点(p2,T2)不仅可以是固相-气相平衡线上的两个点,也可以是液相-汽相平衡线上的两个点。如果平衡蒸汽压可以表示为温度的函数形式,则可以通过该方程得到汽化焓或升华焓。另外,如果已知凝聚相-蒸气相平衡线上某点的升华焓或者汽化焓,则可以通过计算得到平衡线上的其他点。
3. 相变(phase transitions)
热力学体系具有相态的多样性。在不同的宏观约束条件下,物质能够呈现为不同的相态,既可以是单相形态,也可以是多相平衡共存的态。各个相具有显著不同的宏观行为,微观上的行为也明显不同。
对于凝聚态相变和很多其他相变的情形而言,在相变过程中的摩尔熵变与摩尔体积的变化不等于0。然而,在实际应用中仍有许多相变过程的摩尔熵变和摩尔体积变化等于0。
一种常见的相变为一级相变, 其典型特征为如果体系的强度性质发生变化,一旦这些变量或其中之一达到相变发生的临界值时,相变将在宏观上突然发生。一级相变是一种不连续的突变现象,表现出在确定的强度性质的数值时发生,体积、熵、焓等热力学函数值同时发生不连续的但有限的突变。物质的气、液、固态之间的转变都属于这类相变。
另一类相变的特点是热力学函数值的变化是连续的。相变是在强度性质的热力学函数的某一定范围内发生(不是在确定值时发生),而且相变并不表现出体积、熵、焓等的急剧变化, 即它们在相变时是连续的。但其比热容、膨胀系数等性质在相变点附近会发生比较明显的变化,物质的正常状态与超导状态的转变、铁磁铁与顺磁体的转变以及合金的有序与无序的转变等均属于这类相变。此外,还有在相变点时体积、熵、焓连续,而比热容、膨胀系数等性质呈现出不连续但较为有限的突变。在零磁场下超导态金属与正常态金属的转变过程属于这种类型的相变。图3.3中给出了在一级相变与二级相变过程中的化学势(μ)、焓(h)和热容(Cp)变化随温度变化的情况。
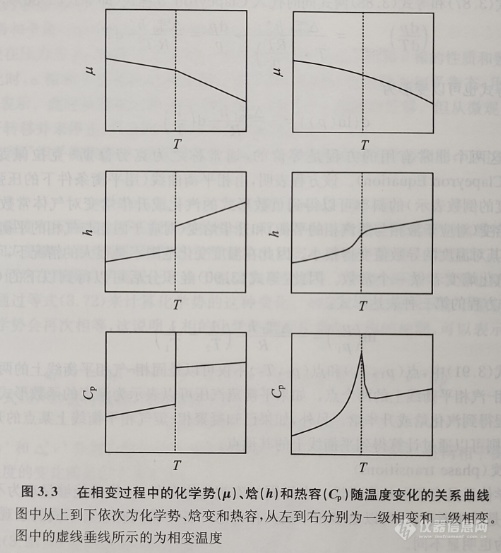
可以用 Clapeyron 方程(式3.86)来确定一级相变平衡曲线的斜率, 但其对连续相变失去意义。Ehrenfest 从理论上导出了二级相变平衡曲线的斜率公式,称为Ehrenfest方程。
Ehrenfest按照级数对相变进行了分类。将满足等式(3.84)假设的相变(在转变过程中,摩尔熵变和摩尔体积的变化均不等于0)称作一级相变(phase transitions of the first order)。在这种情况下,可以认为相变前后的摩尔熵变与摩尔体积的变化分别是摩尔吉布斯自由能(或化学势)对温度与压强的一阶偏导数。因此,可以定义吉布斯自由能对温度与压强的一阶偏导数不为0的一级相变如下:
由于在一级相变的过程中摩尔体积发生了变化,因此可以用热膨胀法测定一级相变。另外,量热法同样也是一种可以用来研究一级相变的非常的工具。由于在相变过程中摩尔熵发生了变化,由此引起了焓变。因此,可以通过量热法测量得到相变热(通常也称为相变潜热)。
当一个相变的吉布斯自由能和摩尔体积变化对温度与压强的一阶偏导数为0时,我们称其为高阶相变(phase transitions of a higher order)。对于一个二级相变来说,根据Ehrenfest的相变理论,其吉布斯自由能对温度与压强的二阶偏导数不等于0,有如下关系式:
因此,对于一个二级相变来说,不存在相变熵(因此也不存在相变焓),同时在相变时摩尔体积也不会发生变化。但是, 在发生二级相变时,体系的热容、热膨胀系数及压缩系数会发生变化。许多所谓的有序-无序相转变(例如铁和镍在居里温度下发生的铁磁到顺磁的相变)都是二级相变的例子。在这些相变过程中,它们的热容随温度的变化曲线往往在转变温度下会表现为一个峰。由于这个峰的形状与字母λ的相似,因此这些相变也通常被称为λ相变。
在通常情况下,对于一个n级相变而言,其吉布斯自由能对温度与压强函数关系的n阶偏导数不为0,而其更低阶的偏导数则为0。
4.玻璃态(the glassy state)及玻璃化转变(the glass transition)
由于目前热分析或量热技术已经广泛应用于玻璃态材料的研究领域,因此在这一节中我们将讨论与玻璃态相关的热力学方面的问题,与此相关的更多的细节问题可以参阅参考文献。
玻璃态是指组成原子不存在结构上的长程有序或平移对称性的一种无定形固体状态,可以看成是保持类玻璃特性的一种固体状态。玻璃态不是物质的一种状态,它是一种无定形的固体结构形式。固态物质分为晶体和非晶体,构成晶体的原子(或离子或分子)具有一定的空间结构(即晶格),晶体具有一定的晶体形状和固定熔点,具有各向异性。玻璃态是一种非晶形态,非晶态是固体中除晶体以外的固体的存在状态。其没有固定的形状和固定的熔点,具有各向同性。它们随着温度的升高逐渐变软,最后呈现熔化状态,外观变软后可加工成各种形状。
在分子尺度上,玻璃态(the glassy state,也称the vitreous state)可以被看做“冻结”(frozen)的液态。在温度发生变化的过程中,相当多的液体可能会出现过冷却(undercooled)现象。当体系被冷却至热力学熔点温度以下时,会形成一种亚稳态的液体(metastable liquid)。如果进一步冷却这种液体,体系往往在一定的过冷温度下开始结晶,即形成稳定的或者亚稳状态的晶体。但在某些情况下,即使在(非常)高的过冷温度下,形成晶体的过程在动力学上仍然是受到限制的。由于此时的(亚稳态)液体的黏度在冷却过程中变大,液体分子的流动速度变得十分缓慢,因此会导致无法形成过冷液体的现象(即无法结晶)。在这种条件下就形成了一种分子构象被冻结的状态,它在更高的温度下会形成典型的液体,通常将这种状态称为玻璃态。但是从机械性质的角度(比如硬度)看,玻璃态具有类似固体的性质。形成玻璃态的过程被称作玻璃化转变过程。通过以上表述我们可以清楚地知道,玻璃态并不是一种平衡状态。
从动力学角度来看,当高分子链段构象重排时,涉及主链上的单键的旋转,键的旋转存在着能垒。当温度在Tg以上时,分子运动导致有足够的能量去克服能垒,最终达到平衡。当温度降低时,分子热运动的能量不足以克服能垒,于是发生分子运动的冻结。由于在转变过程中两个能量状态之间存在着能量差, 这种能量的差异驱动着高聚物玻璃化转变。因此,玻璃化转变现象具有明显的动力学性质,能垒理论从理论上验证了这一点, 它可以很好地解释玻璃化转变中的弛豫现象。但是,根据能垒理论无法从分子结构的角度预测玻璃化转变温度。根据热力学理论很难解释玻璃化转变时复杂的时间依赖性, 而由动力学理论则难以从分子结构角度预测Tg。
通常将过冷液体从类似液体的状态转变为玻璃态的温度称为玻璃化转变温度(glass transition temperature), 用符号Tg表示。玻璃化转变温度与冷却速率有关。如果一种材料冷却得很慢,那么它的玻璃化转变温度将比材料在淬冷条件下的低。这一点与我们在前面一节中所讨论的热力学稳定状态下的相变(无论是一级相变还是更高级的相变)不同。对于这些相变而言,相变温度只与热力学性质有关,而与动力学性质无关。Gibbs和DiMarzio在20世纪60年代末为解释聚合物玻璃化转变而提出玻璃化转变的热力学理论,简称为G-D理论。G-D理论认为:当温度降低时,构象熵随着温度降低而减少;当构象熵降低至零时,物质发生玻璃化转变(构象熵随温度变化)。构象熵包括所有聚合物的构型、位置及取向。通过G-D理论可以成功地解释高聚物玻璃化转变过程中的增塑效应、交联度等问题。
Fox和 Flory提出了玻璃化转变的自由体积理论,他们认为:液体或固体的体积由两部分组成:一部分是被分子占据的体积,称为已占体积;另一部分则是未被占据的体积,称为自由体积。后者以“空穴”的形式分散于整个物质之中,自由体积的存在为分子链通过转动和位移调整构象提供可能性。当高聚物冷却时,自由体积先逐渐减小,当达到某一温度时,自由体积将达到最低值,并维持不变。此时,高聚物呈现玻璃态。因而高聚物的玻璃态可视为等自由状态。自由体积理论采用一个参量——自由体积描述玻璃化转变过程中物性的变化,能够很好地解释玻璃化转变温度附近的黏度和热容随温度的变化关系,但是研究发现:淬火后高聚物在Tg以下,自由体积随着放置时间延长而不断减小。这是自山体积理论的不足之处。
在对一个可以形成玻璃态的材料进行冷却的过程中,如果在高于或者低于玻璃化转变温度时体积是温度的函数并且体积对温度曲线的斜率不相同,则意味着玻璃态的热膨胀系数与过冷液体的不相同。但是,在玻璃化转变温度时、玻璃态材料的体积与过冷液体的体积相同。同样, 在二级相变中也有同样的规律,实际上, 玻璃化转变与二级相变的相似之处远不止如此。等式(3.93)中对二级相变的所有判据也适用于玻璃化转变, 但该过程是准二级相变(pseudo second-order transition), 并不是一个真正意义上的二级相变过程。这主要是因为玻璃态并非一个热力学平衡状态,玻璃化转变温度的数值并不只依赖于其热力学性质,也依赖于其动力学性质(冷却速率)。
在玻璃态形成之后,可以从热力学上定义玻璃化转变温度。此时,玻璃态的体积、熵或焓与过冷液体的相应参数的数值相等。通常情况下,由过冷液体形成玻璃态的过程往往不在一个确定的温度下发生,而是在一个温度范围内发生(反之亦然)。在某些情况下,这个温度的范围变得相当宽。必须通过外推在玻璃态范围与过冷液态范围的体积、熵或焓对温度所得到的曲线的交叉点的方式来确定玻璃化转变温度,交点的温度即为玻璃化转变的温度。
通常用热分析或量热法来研究玻璃化转变,以热容对温度的函数形式来表示测量结果。在玻璃化转变温度附近,热容曲线呈现出不连续性的特征。图3.4给出的是一条典型的甘油的热容对温度曲线。
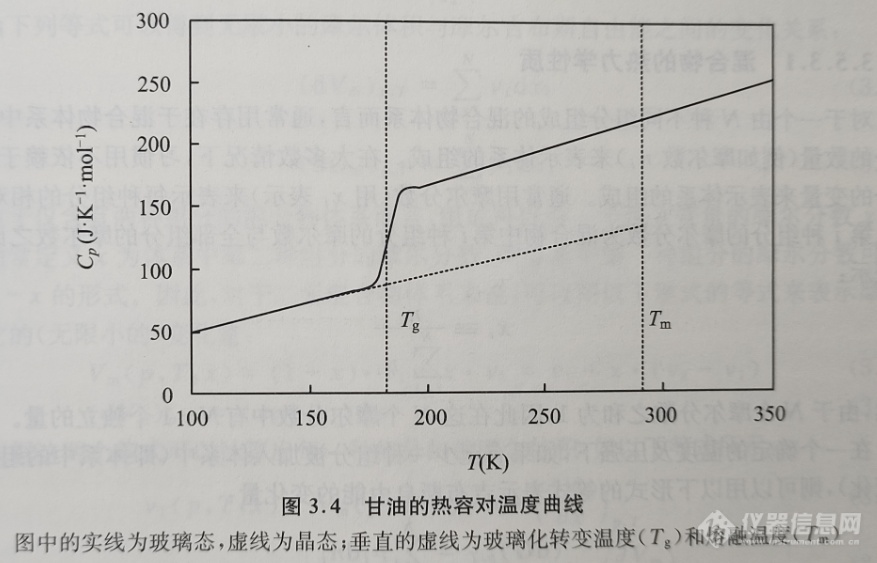
如果玻璃化转变发生在比较宽的温度范围内,则很难直接按照以上的方法由测得的热容对温度曲线确定玻璃化转变温度。通常采用的方法是将在玻璃态与过冷液态的热容数据外推到转变区域, 由实验得到的热容曲线中两条外推曲线的中间值所对应的温度即为玻璃化转变温度;在有些情况下,也可以将由实验得到的热容曲线的拐点温度所对应的温度表示为玻璃化转变温度。显然,通过这两种方式得到的结果都不是本节所定义的严格意义上的玻璃化转变温度。为了得到本节定义的严格的玻璃化转变温度(即准二级相转变温度(pseudo second-order phase transition temperature)), 最好将热容对温度的曲线转化为焓对温度的函数曲线。最后通过外推法找到玻璃态与过冷液态的交叉点,该交点所对应的温度即为玻璃化转变温度(参见图3.5)。
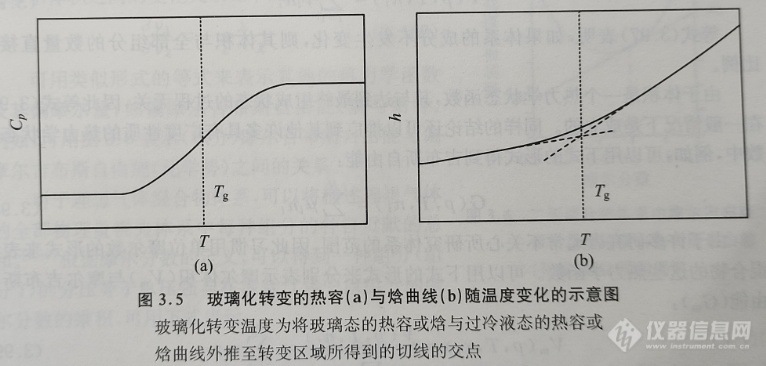
3.5.3 混合物的热力学性质与相图
3.5.3.1 混合物的热力学性质
对于一个由N种不同组分组成的混合物体系而言,通常用存在于混合物体系中的每种成分的数量(例如摩尔数ni)来表示体系的组成。在大多数情况下,习惯用不依赖于体系的成分的变量来表示体系的组成。通常用摩尔分数(用xi表示)来表示每种组分的相对组成,定义第i种组分的摩尔分数为混合物中第i种组分的摩尔数与全部组分的摩尔数之比, 用下式表示:
显然,由于N个摩尔分数之和为1,因此在这N个摩尔分数中有N-1个独立的量。
在一个确定的温度及压强下,如果有至少一种组分被加入体系中(即体系中的组分发生了变化),则可以用以下形式的等式表示吉布斯自由能的变化量:
式中,组分i的化学势μi(或偏摩尔吉布斯自由能)是温度、压强及组成的函数。另外,很多其他热力学广度性质的变化都满足与此相似的等式。例如,可以用下式来表示体积的变化:
组分i的偏摩尔体积vi也是温度、压强及组成的函数。
假设以一种特殊的方式将每种组分加入混合物体系中,并使该混合物体系的组成不发生变化。这意味着在这样一个理想的实验中,组分的偏摩尔体积保持不变。因此,如果对等式(3.96)进行积分,则可以用下式表示其结果:
等式(3.97)表明,如果体系的成分不发生变化,则其体积与全部组分的数量直接成比例。
由于体积是一个热力学状态函数,其与达到最终组成状态的过程无关,因此等式(3.97)在一般情况下是成立的。同样的结论还可以推广到其他许多具有广度性质的热力学状态函数中,例如,可以用下式的形式得到吉布斯自由能:
由于许多研究者通常不关心所研究体系的范围,因此习惯用单位摩尔数的形式来表示混合物的这些热力学函数。可以用下式的形式来分别表示摩尔体积(Vm)与摩尔吉布斯自由能(Gm):
由下列等式可以得到无限小的摩尔体积与摩尔吉布斯自由能之间的变化关系;
对于仅含有两种组分的混合物体系而言,组成可以由一个独立变量的摩尔分数x来表示。通常定义x为体系中第二种组分的摩尔分数, 则体系中第一种组分的摩尔分数可以表示为1-x的形式。因此,对于二元混合物体系来说,可以用以下形式的等式来表示摩尔体积与它的(无限小的)变化量:
根据这两个等式可以计算出每一种组分的偏摩尔体积,如以下等式所示:
分别对等式(3.105)和等式(3,106)以x求偏导,可得到下式:
联立以上两个等式就可以得到著名的Gibbs-Duhem方程式。通过该方程式可知,当压强与温度不发生变化时,由于组分发生的微小变化而导致的偏摩尔体积之间的变化关系如下式所示:
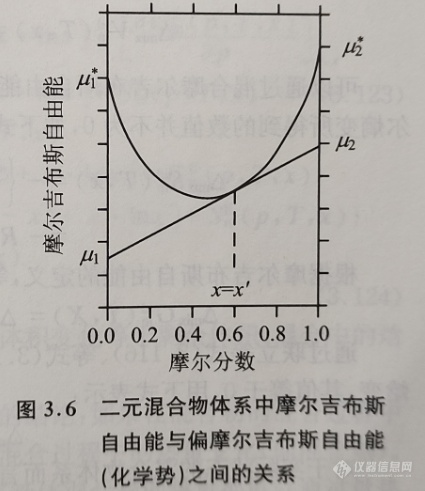
可用类似形式的等式来表示其他的热力学函数及其偏摩尔量,如偏摩尔吉布斯自由能、熵与焓等。例如,可用图3.6表示(积分)摩尔吉布斯自由能与偏摩尔吉布斯自由能(化学势)之间的关系。
对于理想气体混合物体系,可以将描述理想气体的全部物理量视为体系中每种组分的各自贡献的总和。根据摩尔分数的定义,可以得到一种组分(组分i)的分压等于总压强与在混合气体中该组分的摩尔分数的乘积,可用下式表示:
如果混合物中第一种组分和第二种组分的摩尔内能分别为ui(p1,T)和u2(p2,T),则体系中总摩尔内能等于两种组分的各自贡献的总和,可以用下式表示:
对于理想气体而言,内能仅仅与温度有关,而与压强无关。因此,等式(3.111)可以简化为以下等式的形式:
理想气体混合物体系的摩尔吉布斯自由能受组分的影响主要来源于偏摩尔吉布斯自由能或化学势,可以用以下等式表示:
在等式(3.113)中,第一项表示摩尔分数为1-x的第一种纯组分在压强p与温度T下的吉布斯自由能,第二项表示摩尔分数为x的第二种纯组分在相同的压强p与温度T下的吉布斯自由能。显然,第三项表示在混合状态下各组分的吉布斯自由能与未混合状态下的相同组分的吉布斯自由能的差值。因此,这一项是由于理想气体混合物的混合所导致的(摩尔)吉布斯自由能变化(可以记作ΔmixGm的形式)。
对于一个无论是气态、液态还是固态的混合物体系而言,只要它们的吉布斯自由能可以用等式(3.113)来描述,就可以将其视作理想混合物(ideal mixture),即可以认为这类混合物的混合行为与理想气体混合物的混合行为相似。这意味着对于理想状态的混合物而言,可以用下式来表示其混合摩尔吉布斯自由能变化:
通过等式(3.14),可以计算出混合物体系的其他热力学函数。在混合的过程中,可以通过对混合(摩尔)吉布斯自由能对压强的偏微分求得体系的摩尔体积变化。由等式(3.114)可见,对于理想混合物而言,混合摩尔吉布斯自由能的变化与压强的变化无关。因此,在混合过程中体积保持不变,如下式所示:
可以通过混合摩尔吉布斯自由能变化对温度求偏导得到理想混合物在混合过程中的摩尔熵变所得到的数值并不为0,如下式所示:
根据摩尔吉布斯自由能的定义,等式(3.35)可以变换为下式的形式:
通过联立等式(3.116)、等式(3.117)以及等式(3.114),可以求得在混合过程中的摩尔焓变,其值等于0,用下式表示:
对于实际状态的混合物体系而言,通常用理想混合物的摩尔吉布斯自由能与偏离理想状态混合物行为的表达式项的总和来表示体系的摩尔吉布斯自由能。通常称这个偏离项为摩尔剩余吉布斯自由能(excess Gibbs energy,用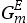 表示)。由于该摩尔剩余吉布斯自由能可能是一个关于压强、温度及组分的函数,因此可以用以下形式的等式来表示实际状态混合物的摩尔吉布斯自由能;
表示)。由于该摩尔剩余吉布斯自由能可能是一个关于压强、温度及组分的函数,因此可以用以下形式的等式来表示实际状态混合物的摩尔吉布斯自由能;
将表示理想状态混合物的摩尔吉布斯自由能的等式(3.114)代入等式(3.119)中,可得下
在等式(3.120)中,前两项表示两种纯组分的各白的贡献量(即未混合状态的吉布斯自由能),第三项表示理想状态的混合物在混合时的吉布斯自由能变化量,第四项是实际状态的混合物在偏离理想状态时的吉布斯自由能变化。可以用下式表示实际状态的混合物在混合过程中的吉布斯白由能变化:
与以上理想混合体系的表示方法类似,如果已知混合过程中的摩尔吉布斯自由能的变化值,则可以计算出在实际的混合过程中的摩尔体积变化、摩尔熵变以及摩尔焓变,如以下等式所示:
由等式(3.122)与等式(3.124)可见,混合过程中的体积变化等于剩余体积,混合中的焓变也就是剩余焓。
由等式(3.124)还可以得出这样的一个特别有意义的结论:如果在混合物的混合过程中压强保持不变,则在混合过程中的焓变(即剩余焓)等于混合过程中的热量变化(即混合热)。这表明,可以通过量热技术测量得到剩余焓。通常,许多液体混合物的热量都可以通过量热仪直接测量得到。对于固态混合物体系而言,只能通过间接的方法测量。例如,可以通过量热法测量得到混合物溶解过程中的热量变化,并可以与纯组分的溶解热进行比较。
3.5.3.2 多组分相图
对于多相体系而言,体系的各相之间的相互转化、新相的形成以及旧相的消失均与其温度、压力、组成密切相关。由实验数据得到的表示相变规律的各种几何图形称为相图。通过这种几何图形可以直观地得到多相体系中的各种聚集状态和它们所处的条件(温度、压力、组成)。相图(phase diagrams)是在给定条件下体系中各相之间建立平衡后热力学函数的轨迹的曲线表达形式, 是表示不同相的稳定区域的图。相图是用来表示材料相的状态和温度及成分关系的综合图形, 其所表示的相的状态是体系的平衡状态, 通常由量热或热分析技术测量得到。由于在前面的内容中已经讨论了仅由一种组分(即一元体系)组成的体系相图,因此在本节中仅讨论二元组分体系(即由两个组分组成的体系)的相图。
1.分相区(region of dcmixing)
可以用等式(3.121)来描述实际状态的混合物体系的吉布斯自由能。对于理想状态的混合物体系,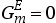 。在整个组成范围内,通过吉布斯自由能对组成(即摩尔比)作图可以得到一条向上凸起的曲线。而当实际状态的混合物体系与理想的混合物存在正偏差(即(
。在整个组成范围内,通过吉布斯自由能对组成(即摩尔比)作图可以得到一条向上凸起的曲线。而当实际状态的混合物体系与理想的混合物存在正偏差(即(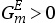 )时,通过其吉布斯自由能对组成(即摩尔比)作图所得到的曲线在整个组成范围内不呈向上凸起状。图3.7(a)中绘制了一条在一定温度T=Θ(和一定压力)下的摩尔吉布斯自由能与摩尔组成之间的函数关系曲线。对于组成为X′
)时,通过其吉布斯自由能对组成(即摩尔比)作图所得到的曲线在整个组成范围内不呈向上凸起状。图3.7(a)中绘制了一条在一定温度T=Θ(和一定压力)下的摩尔吉布斯自由能与摩尔组成之间的函数关系曲线。对于组成为X′
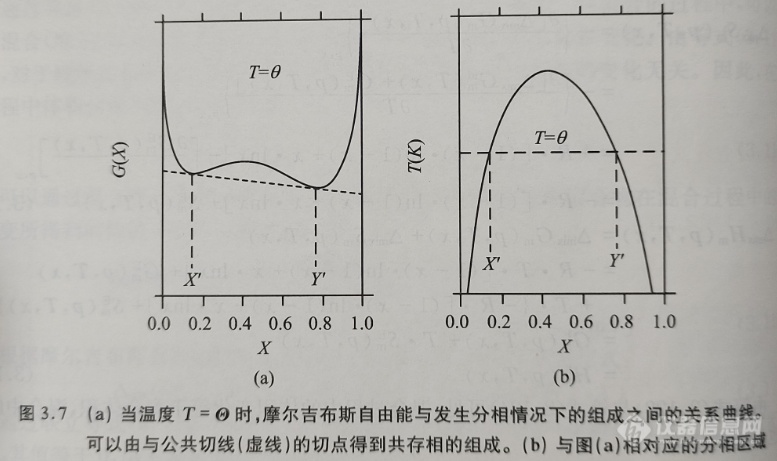
由于分相体系的吉布斯自由能低于混合体系的吉布斯自由能,因此分相体系在热力学上是稳定的。在其他的温度下,X′和Y′的值也会发生改变。通过数据点(T,X′)和(T,Y′)可以绘制得到温度随组成的变化关系曲线,通常称该曲线为双结线(binodal)。由双结线包围的区域通常称为分相区(region of demixing)或不相混溶区(miscibility gap),典型的分相区如图3.7(b)所示。在这种情况下,组分的相互混溶性随着温度的升高而增加。在曲线中温度随组成的变化出现了最大值,通常称该最大值为临界点(critical point),与此相对应的温度称为临界温度(critical temperature)。然而,在体系中还存在着组分的相互混溶性随着温度升高而降低的分相区。由于这些分相区在双结点处存在最小值, 因此该临界点较低。由于封闭的分相区域(closed regions of demixing)具有低临界点和高临界点,因此这意味着均匀混合物在冷却时可以分为两相,并且在进一步冷却时可能会出现再次混合的现象。
图3.7(a)中,当温度T=Θ时,平衡状态的组成为x=X′和x=Y′。可以绘制出在这两个值(X′1和μ2(也可参见图3.6)。可以用以下两式来表示平衡条件:
注意,以上这两个平衡条件必须同时满足。由图3.7可见,这些平衡条件相当于
2.热力学相图分析
如果纯组分的热力学性质和热力学混合性质已知,可以根据以上所述平衡条件计算相图。此外,从已知的(实验确定的)相图来推导热力学混合特性也是可以实现的,这个过程被称为热力学相图分析。
热力学相图分析是一种拟合方法,它利用了剩余热力学函数,在拟合过程中需要根据实际来调整的参数是这些函数的系数。
热力学和图分析的结果,即剩余热力学函数的数学表达式,对应于与热力学相一致的计算相图,据其还可以很好地再现实验相图。因此, 热力学相图分析是一种可靠地拟合实验相图数据的方法,也是测试这些数据内在一致性的手段。如果需要从二元相图的数据估计三元和高阶相图,则首先需要进行热力学相图分析。
有关热力学相图分析的更深入的信息,可以参阅参考文献[14]。
3.5.4 化学反应的热力学平衡
3.5.4.1反应吉布斯自由能、反应熵和反应焓
化学反应是一种或多种组分的分子结构(这些组分称为反应物)变化的过程,所形成的新组分称为产物。反应物中一部分化学键断裂,同时在产物中有新的化学键形成。一般使用化学计量方程式来描述化学反应,例如:
这意味着1 mol N2与3mol H2反应形成2mol NH3。原则上如果改变温度和分压,反应也可以在逆方向上进行:
对于特定的温度,可以实现这样的分压条件,使得NH3既不形成也不分解。在这种情况下,体系处于平衡状态:
在恒定的压力和温度下,自发(不可逆)过程的特征是体系吉布斯自由能降低。因此,当需要探究组分之间是否可以发生自发反应时,必须确定由化学反应引起的吉布斯自由能的变化。假设反应物A和B之间发生化学反应,生成了产物C和D,如下式所示:
值得注意的是,在以上方程式中,系数vA和vB均带绝对值符号。这是因为系数vA和vB(即反应物的系数)被定义为负值,而系数vC和vD(即产物的系数)被定义为正值。使用符号ξ表示反应程度,为0到1之间的数值。若ξ=0,表明体系中只有反应物;若ξ=1,则表示体系中只存在产物。当反应程度dξ发生微小的变化时,将导致反应物数量的减少(即A和B 的量分别减去dnA=vAdξ和dnB=vBdξ)和产物数量的增加(即C和D量分别加上 dnC= vCdξ 和dnD=vDdξ)。
由于吉布斯自由能属于状态函数,因此由反应程度变化所引起的吉布斯自由能的变化即为反应吉布斯自由能变化值,在反应过程中,如果A和B按照dnA和dnB摩尔数从体系中可逆地除去,则dnC和dnD摩尔数的C和D将可逆地被加入体系中。因此,根据等式(3.73),在恒定温度和压力下,可以由下式给出化学反应吉布斯自由能的变化:
或者
该等式可以改写为更加通用的表达式:
式中,求和式涵盖了反应中所有反应物和产物。如果吉布斯自由能处于其最低值,则反应达到平衡状态,即
通常定义反应吉布斯自由能为化学势与化学计量系数的乘积总和,为方便起见,该自由能用符号ΔrG表示:
原则上,由于(在恒定的温度和压力下)吉布斯自由能趋于最小值,因此若反应的吉布斯自白由能为负值,则反应趋于向化学计量方程式的右侧进行;若吉布斯自由能变化值为正值,则反应趋于向化学计量方程式的左侧进行。
可以由下式得出反应吉布斯自由能对于温度的依赖性,即反应的熵;
结合等式(3.156)和等式(3.157)可推导出反应焓,如下式所示:
反应焓是一个重要的物理量,对于恒定压力下的过程,反应焓等于反应热,可通过量热技术实验获得反应焓。
3.5.4.2 化学平衡
反应吉布斯自由能的定义如等式(3.155)所示。如果反应涉及混合物(固体、液体或气体)和/或纯气体,那么等式(3.155)中的化学势是混合物组成的函数。由于在反应期间,混合物的组成和/或分压可能改变,因此反应吉布斯自由能也可能会发生改变。若反应的吉布斯自由能为零,则达到了平衡态。
可以按照此标准来计算分解过程的化学平衡,这种情形在热重分析实验中十分常见。
例如,对于以下的碳酸钙分解反应的吉布斯自由能:
其反应吉布斯自由能ΔrG可以表示为
上式中,碳酸钙(方解石)和氧化钙都处于固体状态, 并且这两种组分之间不形成混合态固相。因此,可用星号标记这些组分化学势,表明它们是纯组分的特性。气态二氧化碳的化学势是二氧化碳分压的函数。若假设气体为理想气体,则可用等式(3.75)描述二氧化碳的化学势。考虑到上述因素,可以将等式(3.160)重新改写为
通常物理量ΔrG0为反应的标准吉布斯自由能。反应的标准吉布斯自由能定义为:在没有形成混合物(即所有反应物和产物都是纯的),并且所有分压都等于标准压力(通常为1 bar)的(假想)情况下反应的吉布斯自由能。虽然反应的标准吉布斯自由能原则上是温度的函数,但根据定义知它不是压力的函数。
当涉及更多气体组分参与反应时,等式(3.163)变形为
式中的求和部分涵盖反应中涉及的所有气态组分。
当反应的吉布斯自由能等于零时,反应处于化学平衡状态。因此,可用下式的形式表示平衡压力pe(CO2)与反应的标准吉布斯自由能之间的关系:
下式中给出了从生成吉布斯自由能(ΔfG)计算反应吉布斯自由能的过程:
等式(3.166)中的CaO、CO2、CaCO3的生成吉布斯自由能的数值可以通过查表得到。因此,可以将等式(3.165)重新改写为下式的形式:
根据上式可以计算得到不同温度下的二氧化碳的平衡分压。如果二氧化碳的分压大于平衡压力,那么碳酸钙是稳定的(或氧化钙将会被转化为碳酸钙);如果二氧化碳的分压低于平衡压力,则可以观察到碳酸钙被分解成氧化钙。同时,在热重分析实验中可以观察到质量损失(因为二氧化碳气体将离开样品容器)。若这样的实验在常压下的纯二氧化碳气氛中进行,则当温度高于1169K时碳酸钙会发生分解;若此实验在大气条件下(即空气中)进行,当总压为1bar时,二氧化碳分压p(CO2)=3.3×10-4bar,则在当温度高于802K时碳酸钙会发生分解。在实际应用中可以通过以上的热力学分析方法来与实际的实验结果进行对比。
产品货期: 2天
整机质保期: 3年
培训服务: 安装调试现场免费培训
安装调试时间: 到货后2天内
电话支持响应时间: 2小时内
是否提供维保合同: 是
维修响应时间: 1天内
节假日是否提供上门服务: 是
核心零部件货期: 2天
核心零部支持时间: 20年
是否支持上门巡检: 是
是否提供预防性维护计划: 是
是否提供期间核查方案: 是
是否提供免费应用支持: 是
是否提供付费应用支持: 是
是否提供线上售后平台: 是
维修付款方式: 先维修后付款
基本维修资料公开: 技术参数
无理由退换货: 不支持
其他: 用心服务,尽最大努力做到客户的满意。
中航时代差示扫描量热仪DSC差示扫描量热仪-DSC差示扫描量热分析仪的工作原理介绍
差示扫描量热仪DSC差示扫描量热仪-DSC差示扫描量热分析仪的使用方法?
中航时代DSC差示扫描量热仪-DSC差示扫描量热分析仪多少钱一台?
差示扫描量热仪DSC差示扫描量热仪-DSC差示扫描量热分析仪可以检测什么?
差示扫描量热仪DSC差示扫描量热仪-DSC差示扫描量热分析仪使用的注意事项?
中航时代DSC差示扫描量热仪-DSC差示扫描量热分析仪的说明书有吗?
中航时代差示扫描量热仪DSC差示扫描量热仪-DSC差示扫描量热分析仪的操作规程有吗?
中航时代差示扫描量热仪DSC差示扫描量热仪-DSC差示扫描量热分析仪报价含票含运吗?
中航时代DSC差示扫描量热仪-DSC差示扫描量热分析仪有现货吗?































