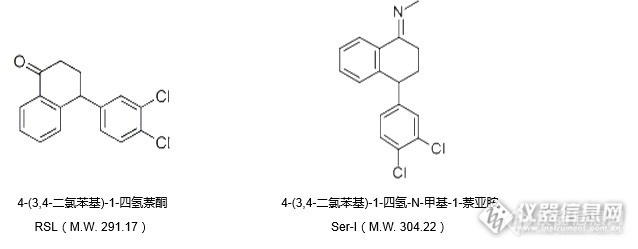
[align=center]萘酮与中间体杂质I的分离[/align]根据客户提出的依赖分析需求,实验室对以下结构的萘酮(RSL)及其中间体杂质I(Ser-I)进行分离尝试。[align=center][img=,638,249]http://ng1.17img.cn/bbsfiles/images/2017/06/201706210849_01_2222981_3.png[/img][/align][align=center][img=,690,226]http://ng1.17img.cn/bbsfiles/images/2017/06/201706210850_01_2222981_3.png[/img][/align]注:在客户给出的数据文件中,RSL命名为萘酮,Ser-I命名为中间体I;在加磷酸体系中,中间体I先出峰,不加磷酸体系中,萘酮先出峰。由于萘酮(RSL)与中间体I(Ser-I)在水相中会发生结构转换现象,因此我们在无水条件下开展实验。使用资生堂疏水性与表面极性得到良好平衡的反相色谱柱CAPCELL PAK C18 MG S5 4.6 mm i.d. × 250 mm进行分析,同时对柱温进行优化,结果如图1所示。[align=center][img=,690,280]http://ng1.17img.cn/bbsfiles/images/2017/06/201706210849_02_2222981_3.png[/img][/align][img=,581,198]http://ng1.17img.cn/bbsfiles/images/2017/06/201706210849_04_2222981_3.png[/img]图2、图3分别为萘酮和杂质I的光谱图。[align=center][img=,690,271]http://ng1.17img.cn/bbsfiles/images/2017/06/201706210849_03_2222981_3.png[/img][/align]由图1可知,在萘酮的分析中,柱温越高其保留时间越短。同时发现在萘酮与杂质I之间出现一较明显倒峰。由图2、图3决定检测波长,由于流动相中添加了三乙胺,会对短波长检测产生一定干扰,因此建议在254nm或者288nm进行检测(本实验选择254nm)。我们对图1中倒峰的来源进行了多方排查,最终发现该实验体系中不得引入任何水,建议客户使用的所有实验容器必须烘干,并且需将洗针液更换为纯有机相。排除水干扰后分析对比结果如图4所示。[align=center][img=,638,363]http://ng1.17img.cn/bbsfiles/images/2017/06/201706210849_05_2222981_3.png[/img][/align]同时,为进一步延长保留时间,我们也尝试使用了资生堂键合金刚烷基团的高表面极性色谱柱CAPCELL PAK ADME S5 4.6 mm i.d. × 250 mm进行分析,所得结果如图5所示,相较于MG色谱柱,ADME色谱柱能够得到更强保留。[align=center][img=,616,304]http://ng1.17img.cn/bbsfiles/images/2017/06/201706210849_06_2222981_3.png[/img][/align]
怎样利用碳谱测活泼中间体?谢谢
医药中间体是一种用于药品合成的化工原料或化工产品。这种化工产品,不需要药品的生产许可证,在普通的化工厂即可生产,只要达到一些的级别,即可用于药品的合成。中间包括化学试剂的各个分类,比如铵苯镝胍铁酮脂唑铈等。中间体非常大的部分属于半成品,属于工艺中间的,必须经过一定工艺处理的初产物,也就是还属于工业材料,不是最终产品。医药中间体属精细化工产品,生产医药中间体目前已成为国际化工界的一大产业。同时作为半成品,医药中间体属精细化工产品,生产医药中间体目前已成为国际化工界的一大产业。常用的医药中间体有,2-乙基苯骈呋喃 3,5-二溴-4-羟基苯甲酸 4-甲基-3-硝基苯甲醚 普瑞巴林 烯炔醇 米托坦 N-氯代酞酰亚胺 6-氟-3-哌啶-4-基-1,2-苯并异唑盐酸盐 等,就生产而言,国内大多数企业海处于起步阶段,和欧美等药业巨头相比还有一定差距。与此同时,医药中间体与原料药相比,生产中间体利润率偏低,而原料药与医药中间体的生产过程又相似,因此,部分企业已不仅仅生产中间体,还利用自身优势,开始生产原料药。如何改进医药中间体的生产工艺,降低生产成本,以及寻找合适的路线生产原料药,无疑会获得更大的利润。医药中间体产品剖析一般采用红外光谱(FTIR)、核磁共振(1H NMR)、质谱(MS)、X衍射分析(XRD)、ICP-MS、X荧光光谱分析、离子色谱分析等手段。通过这些测试手段可以很好的解析医药中间体的配方,对医药中间体中的成分作用有详细的了解,更方便各个企业进行研发,把握市场动态。
制剂的中间物料如压片前的混合粉末,是叫中间体还是中间产品呢?原料药的中间产物呢?看有的资料写中间产品有的写中间体,区别大吗?
(求助)阿托伐他汀中间体的合成文献
在制备阿托伐他汀钙中间体A7过程中,色谱发现了有个含量极高的异构体,但不清楚具体是什么。现在想把这个异构体分离出来,我们已经通过结晶去掉了成品,现在通过减压蒸馏脱掉溶剂,里面异构体含量约70~80%,但没办法得到固体。现在问题是:通过什么方法能将成品与异构体分离并对异构体进行提纯呢?
有个疑问,关于生产中间体的测试项目和成品测试项目一样,而中间体生产后只是经过灌装的步骤,是否可以把中间体测试结果做为成品的测试结果,然后再加做一个微生物的测试。不知道是不是概念不清,请大家帮我理理。谢谢!
国内注册CTD格式中有一条:列出已分离的中间体的质量控制标准,包括项目、方法和限度,并提供必要的方法学验证资料。对于已分离的中间体,怎么理解?
我目前在做咔唑合成 氯乙基咔唑,最终产物是乙烯基咔唑咔唑是买的原料基本上厂商的分析方法是气相 咔唑的沸点很高 354实验室没有耐温这么高的柱子我现在就是用液相分析 乙烯基咔唑的分析方法240nm 乙腈+水=70+30 0.1ml/min ODS-3出峰还不错,针对方法未进行验证现在纳闷的是像氯乙基咔唑这种中间体一般都买不到标样这个情况如何分析像氯乙基咔唑这类是否是属于盐类呢 、
在很多文献中,可以见到利用核磁共振技术监测反应的进行,观测可能的中间体.众所周知,反应中间体的浓度通常是很低的,用一般方法不易观测到.请问用核磁进行中间体检测,浓度下限可以到多少?如何从反应体系的复杂信号中将中间体的信息剥离出来?
他达拉菲中间体厂家直销19953321917
http://www.3158.com/upfiles4/2010/08/24/15/06/07/6599bb85.jpg请教各位版友,用液相法分析甲磺酸伊马替尼(见上式),其中有个中间体是没有与甲磺酸成盐的成分。在液相的溶液条件下(水相pH2.5),能把这两种物质分开吗?甲磺酸伊马替尼是可以水解的吧?这个中间体和甲磺酸依马替尼会生成同一种物质吗?要是这样的话,那这个中间体就没法分析了吧。谢谢各位版友!
反应中间体是一个有机Li试剂,只能在-78度以下存在。这样的中间体如果想用NMR来检测,怎么一个检测法?除了低温NMR以外,有没有其他的好方法?谢谢大家!
小弟以前在学校图书馆看到一本 有机中间体的制备与合成手册 是本红色封面的,现在没看到外面有卖的,不知道哪为GGJJ有没有电子版的.如果能传上来的话.不胜感激!
最近在消解一个药物中间体,准备测试里面的Fe, Al等。在消解时出现了下面的情况:消解后是澄清的,加水变浑浊,再加酸又澄清,另外不存在酸少的问题,试过0.1 g样品加15 mL硝酸的情况,温度最高设置过230度,时间设置20 min。以前没有接触过药物中间体的消解,不知在做药物中间体消解时有哪些需要注意的地方?谢谢。
作者:郑伟 孙光等题目:色满酮类化合物的合成及其抗炎活性的研究期刊:精细化工中间体年卷期:2009,39(1)页码:30-33中文-万方
老师:今天我做了一个头孢中间体,分别用DMSO、D2O做溶剂,两张谱图相差很大,是溶剂效应吗?谱图是扫描的,效果不太好。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=49432]谱图及结构式[/url]
我曾在各种各样的企业待过。有一家客户是欧美等发达国家的,他们要求原料、中间体、中控方法都必须进行方法验证。而我们国内注册的标准中,只对原料分析方法验证有简单的要求,对中空分析方法、中间体分析方法并没有要求。大家一起来讨论讨论,发表各自的观点。
在检测注射用甲磺酸左氧氟沙星含量时:中间体检测时用紫外检测,成品用的是高效液相检测,之前中间体用原料做工作对照,对照品A值一般在所配浓度下一般为0.39左右,F值在1.26左右,成品没什么问题,可是现在用中检所的对照品,中间体测定时对照品在相同浓度下A值才有0.35左右,这样的话F值就接近1.3了,结果含量就会高很多甚至不合格,但是成品上液相时又是合格的,也就是说对照品应该没问题,是不是中检所的对照品不适合用紫外分光光度法来检测呢?请各位帮忙分析分析,谢谢!!!补充:在这之前,该产品中间体检测都是用中检所的对照品上高效液相测定的,为了节省时间改用了紫外分光光度法检测工艺没改,液相测出来接过肯定会更精确些,但是奇怪的是,同样的对照品中间体检测超标,成品检测合格,不过之前用原料做工作对照检中间体时,成品标示量一般在100%以下,用中检所对照品检测中间体,成品标示量一般在105%,规定上限是110%没我是在想我们的中间体检测方法是不是不太合理制药工艺中没有纯化过程的,但是原料工作对照77.54%,中检所对照品97.3%,都是根据所需浓度换算好了再配制检测的,我是想应该里面所含杂质吸收影响就不明显了吧?您觉得呢?
过期 长时间存放的 化工 原料 谁哪里呢有呢 助剂 油漆 中间体 等等化工原料
早在2010年的5月,欧盟委员会、欧盟成员国的监管机构及ECHA进行了商定,对REACH法规下的中间体的含义进行了澄清,并以非官方的“草案指南”的形式对澄清进行了发表,最终其将在2010年11月30日后并入ECHA关于中间体的指南中。 虽然,该草案指南没有法律约束力,但是按照以往惯例,ECHA的指南通常都考虑了执法目的并在可能的情况下产生法律效力。特别是作为中间体而言,其有可能落入REACH监管范围外(如非分离的中间体),也可能落入REACH法规的监管范围内(现场分离中间体及可转移的中间体)。因此,该指南文件更是可能对业界产生深远的影响。 为此,欧盟的法律公司Field Fisher Waterhouse对中间体的定义进行了独立分析,该分析是遵循典型的欧洲法院的做法进行的,即:强调字面的释义,由系统及技术解释的支持。 对具体分析结果简述如下: 一、对中间体的字面和系统/技术的释义 REACH法规的第3(15)条已对“中间体”的定义进行了明确、清晰的解释。在该条款中,指出了“中间体”指为“将一种物质转化为另外一种物质所进行的化学反应中制造、消耗或者使用的物质”。Field Fisher Waterhouse认为,字面上看该定义中确定物质是否为“中间体”的关键是是否故意进行了转化,而不论其最终的用途:制造、消耗或者使用。 而从系统/技术方面而言,对中间体定义的阐述需要考虑到两方面:REACH法规的目的;豁免后隐藏的理由。早在REACH制定之初,就考虑到了作为制造商、进口商的一方和作为欧盟及成员国的另一方在实际操作中有许多的困难。为此,欧盟有意减少中间体的注册要求。因此,中间体是在化学过程中转换为另一种物质的物质:不论整个转化的目的; 不论转化在制造过程中是即时的步骤,或是连串的步骤; 不论最终产物在同一过程中是否继续使用,亦或是直接在混合物或物品中销售,或直接市售; 不论该物质是否在某些情况下被视为中间体,而在另外一些情况下不被视为中间体; 不论由该中间体转变而来的物质是否受REACH法规的豁免。 当然,关于REACH法规下的中间体的释义还需要具体案例来具体分析,但是有些情况下,某些物质可以肯定其一定不是中间体,如催化剂。 二、ECHA对于中间体概念的最新发展 ECHA的最新的“草案指南”中,其对中间体的定义新增的最显著的几点为:可分离中间体转变为其他物质必须出现在中间体制造的随后的步骤中; 化学过程的主要目的是一种物质转变为另外一种物质,而非获得其他的功能; REACH法规附件V第3、4条予以豁免的物质不能作为中间体,因为其主要是用以提供特殊功能及相应的理化性质。 对于ECHA进行的释义的新发展,Field Fisher Waterhouse认为其对之前REACH法规中的定义加入了新释义,其并未基于之前定义的字面及系统/技术的释义。并且,其缩小了中间体的定义范围,可能导致很多原本属于中间体范围的物质不再落入该范围中。这应引起业界的关注。信息来源:技术壁垒资源网
在GMP认证过程中中间体化验需要做那些工作呢?化验室内都需要有那些标识呢?希望各位大师帮一下忙,谢拉!
想自己创建一个医药中间体实验室,公斤级的.希望大家给指点指点.150平左右.
如题,三类化药中间体,需要做降解实验吗?如果需要,降解到什么程度?有官方的指导原则吗?如果有,请指出。谢谢。
请问染料中间体偶合值是指什么?怎么测定啊领导需要测定染料中间体的偶合值,我连它是什么,表示什么都不知道啊我查了资料时这样理解的:可发生重氮化反应的芳伯胺含量。哪位大侠指点一下,着急啊
[sub]点击链接查看更多:[url]https://www.woyaoce.cn/service/info-37631.html[/url]?[font=&][size=16px][color=#333333]服务背景[/color][/size][/font][font=&][color=#333333][/color][/font][size=16px] [font=黑体, SimHei]医药中间体,实际上是一些用于药品合成工艺过程中的一些化工原料或化工产品。,医药检测领域的主要服务范围有药包材相容性研究、药包材密封性验证、药品测试、中药测试、药品成分分析、稳定性试验研究等。[/font][/size][font=&][size=16px][color=#333333]检测内容[/color][/size][/font][font=&][color=#333333][/color][/font][font=黑体, SimHei]纯度、成分鉴定、原料药结构分析、成分配比等[/font][font=黑体, SimHei][size=16px]定性定量分析、解析医药中间体的配方等[/size][/font][font=黑体, SimHei][size=16px]方法学验证:原料药方法学验证、医药中间体方法学验证、药品有关物质检测、药品有关物质鉴定、药品杂质鉴定、药品杂质结构检测、杂质鉴定、结构鉴定;[/size][/font][font=黑体, SimHei][size=16px]分子量:分子量分布、分子量检测、蛋白质分子量测定;[/size][/font][font=黑体, SimHei][size=16px]采用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]分析、红外光谱(FTIR)、质谱(MS)、X衍射分析(XRD)、核磁共振(1H NMR)、[url=https://insevent.instrument.com.cn/t/yp][color=#3333ff]ICP-MS[/color][/url]、X荧光光谱分析等方式。[/size][/font][font=&][size=16px][color=#333333]检测标准[/color][/size][/font][font=&][color=#333333][/color][/font][/sub][table][tr][td]产品名称[/td][td]检测项目[/td][td]检测标准[/td][/tr][tr][td]药包材[/td][td]相容性研究[/td][td]YBB00032005-2015[/td][/tr][/table]
手上正在探究几个中间体的分析方法,现将其中的一个或几个问题,跟大家分享下,希望对大家有所帮助(谱图稍后传上)样品:中间体(合成提纯后样品,没进行定量分析,理论认为纯度较高)分析方法一、流动相:乙腈+水=60+40 流速:1.0ml/min 波长:280nm 恒流 岛津LC-10AD 无在线脱气装置流动相超声,脱气,柱平衡后进样,压力72Kgf/cm2见谱图(乙腈)分析方法二、流动相:甲醇+水=70+30 流速:1.0ml/min 波长:280nm 恒流 岛津LC-10AD 无在线脱气装置流动相超声,脱气,柱平衡后进样,压力115Kgf/cm2见谱图(甲醇)根据谱图情况分析方法的优越性,极其考虑过程:拿到样品,先了解样品的一些理化性质,稳定性,然后做个UV确定下最大吸收波长(因样品不是太纯,测的有两个)备注:大家注意下最大吸收波长和合适吸收波长这两个概念,合适吸收波长一般在最大吸收波长上下20nm左右,一般我们用的波长都是合适吸收波长,根据谱图进行调节;一般针对原药和含量超低的选用最大吸收波长。确定波长后,就要选择流动相和色谱柱。80%的化合物都可以用C18 这里我就不多说了流动相,首先选择乙腈,因为乙腈的洗脱能力强,出峰较快,能较块的知道效果,然后在进行实际情况微调。我选择乙腈和水做流动相,进行微调后,发现分离效果不错,但是峰形却相当不好看,中等程度的拖尾。见谱图(乙腈)通过加磷酸,不断调试PH,但对峰形没有多大影响。后改用甲醇水做流动相,出峰效果非常好,见谱图(甲醇)至于用甲醇代替乙腈,为什么能取得如此的效果,我是这样理解的,因为为了达到同样的保留时间,不更改其他条件的情况下,提高有机相的浓度,有利于峰形改善。现在确定分析方法二为本产品的分析方法。补充:UV测得两个波长,一个是270nm,一个280nm,用270nm定为检测波长时,发现主峰钱有个峰,峰高较高,影响美感,而用280nm则不会有此情况,所以确定为280nm。见图(E-3)http://ng1.17img.cn/bbsfiles/images/2011/09/201109061800_314650_1638724_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109061801_314651_1638724_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/09/201109061801_314652_1638724_3.jpg
如题。原料药的中间体的检测标准是如何建立的?对于制剂和原料药成品,都有相应的药典标准。那么中间体的检测标准依据什么呢?又是如何建立的呢?畅所欲言,经验交流~
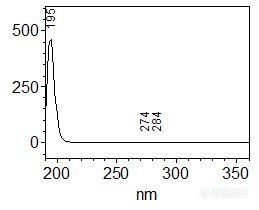
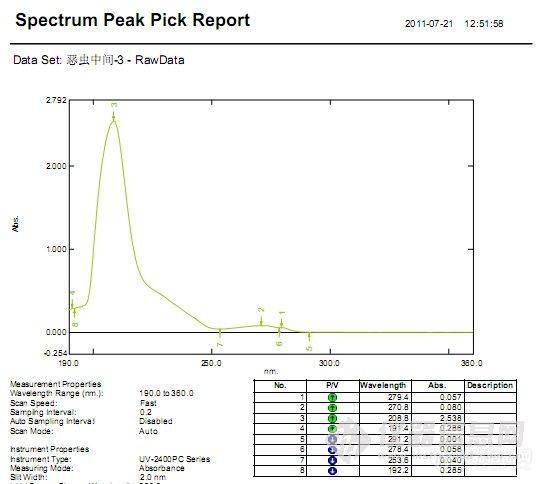
[align=center][b]左卡尼汀中间体及成品的分析——PDA及NQAD检测器对比[/b][/align]客户提供已知结构中间体2和中间体3,以及成品左卡尼汀单标样品,希望能够建立中间体液相分析检测方法。由中间体结构式可知其紫外吸收较弱,因此首先使用二极管阵列检测器——PDA,对其紫外吸收进行确认。紫外吸收光谱图如图1和图2所示,二者均为短波长吸收,最大吸收波长分别为195 nm和194 nm,最终选择的紫外检测波长为195 nm。[align=center][img=,259,210]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251523394585_463_2222981_3.jpg!w259x210.jpg[/img] [img=,252,204]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251523440193_5542_2222981_3.jpg!w252x204.jpg[/img][/align][align=center] 图1 中间体2紫外吸收光谱图 图2 中间体3紫外吸收光谱图[/align]接下来进行液相方法的建立。考虑到中间体的极性较强,故首先使用具有超高表面极性的反相柱CAPCELL PAK ADME,在100%水相条件下进行保留尝试,缓冲盐选择在低波长干扰较小的高氯酸钠体系。分析结果如图3所示,两中间体在反相系色谱柱上均无法得到保留,反相系色谱柱不适用于该项目分析。[align=center][img=,449,258]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251530523604_5470_2222981_3.jpg!w449x258.jpg[/img][/align][align=center]图3 CAPCELL PAK ADME色谱柱分析结果[/align][img=,451,170]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251530520833_9064_2222981_3.jpg!w451x170.jpg[/img]接下来,考虑利用阳离子交换模式和亲水性相互作用模式进行分析,以期得到良好保留和峰形。经过多方条件调整后,最终在亲水性相互作用色谱柱PC HILIC上得到良好保留结果,中间体分析谱图及放大图分别如图4-7所示。中间体在高氯酸钠体系下得到良好峰形,同时与死时间附近杂质峰取得了良好分离。应客户要求对其杂质进行了积分,积分表分别如表1-2所示(软件自动积分结果)。[align=center][img=,389,236]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251532088480_8928_2222981_3.jpg!w389x236.jpg[/img][/align][align=center]图4 中间体2分析结果[/align][align=center][img=,389,235]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251532091260_9820_2222981_3.jpg!w389x235.jpg[/img][/align][align=center]图5 中间体2分析结果放大图[/align][align=center] [/align][align=center]表1 中间体2分析结果积分表[/align][align=center][img=,624,541]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251532085430_4956_2222981_3.jpg!w624x541.jpg[/img][/align][align=center][/align][align=center][img=,509,298]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251536544585_463_2222981_3.jpg!w509x298.jpg[/img][/align][align=center]图6 中间体3分析结果[/align][align=center][img=,494,302]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251536551365_8367_2222981_3.jpg!w494x302.jpg[/img][/align][align=center]图7 中间体3分析结果放大图[/align][align=center] [/align][align=center]表2 中间体3分析结果积分表[/align][align=center][img=,562,566]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251537328944_2982_2222981_3.jpg!w562x566.jpg[/img][/align][align=center][/align][align=left][img=,549,237]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251538004262_3029_2222981_3.jpg!w549x237.jpg[/img][/align][align=left][/align][align=left]同时,我们也使用PC HILIC色谱柱进行了两中间体的共同分析,以期得到二者的基线分离结果,简化分析过程。然而在HILIC模式下,由于乙腈比例较高,且中间体自身需要在短波长下检测,因此可选的缓冲盐浓度及种类均有限;水相分别尝试使用0.1%磷酸溶液、20 mmol/L磷酸二氢钾溶液(磷酸调pH 2.5)、20 mmol/L磷酸二氢铵溶液(磷酸调pH 2.5)及不同浓度高氯酸钠溶液,结果均无法得到两中间体的分离;且在盐浓度不足时,中间体由于自身的季铵盐结构易产生吸附作用,很难得到良好峰形,典型谱图如图8所示。[/align][align=left][/align][align=center][img=,543,285]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251538324635_1675_2222981_3.jpg!w543x285.jpg[/img][/align][align=center]图8 0.1%磷酸条件PC HILIC分析结果[/align][align=center][/align][align=left]此外对柱温进行筛选,温度升高时保留时间有缩短趋势,但未见二者出现明显分离趋势,结果如图9所示。[/align][align=left][/align][align=center][img=,541,385]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251539139335_5664_2222981_3.jpg!w541x385.jpg[/img][/align][align=center]图9 不同柱温分析结果[/align][align=center][/align][align=left]考虑到中间体的整体紫外吸收较弱,应客户要求,使用高灵敏度气溶胶型检测器NQAD进行了分析对比。[/align][align=left]由于气溶胶型检测器NQAD要求流动相必须为挥发性盐,因此将水相中的高氯酸钠更换为50 mmol/L甲酸铵溶液(+0.1%甲酸)进行分析,中间体2、中间体3、左卡尼汀共同分析结果如图10-12所示。由于5 mg/mL样品浓度较大,在NQAD检测器上过载,出现信号平头峰或裂峰现象,把浓度稀释10倍后可得到正常峰形。但即使在大浓度样品分析中,未见死时间附近出现明显杂质峰,推测原因可能为杂质分子量较小,为挥发性杂质,无法在气溶胶型检测器上进行良好检出。此外,能够看到中间体在NQAD检测器上均出现多个明显色谱峰。[/align][align=left][/align][align=center][img=,674,407]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251540311232_3541_2222981_3.jpg!w674x407.jpg[/img][/align][align=center]图10 中间体2在NQAD检测器分析结果[/align][align=center][img=,670,402]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251550168795_4568_2222981_3.jpg!w670x402.jpg[/img][/align][align=center]图11 中间体3在NQAD检测器分析结果[/align][align=center][img=,677,395]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251550176905_730_2222981_3.jpg!w677x395.jpg[/img][/align][align=center]图12 左卡尼汀在NQAD上分析结果[/align][align=left]由于使用NQAD进行检测时,中间体均出现溶出多个色谱峰现象,怀疑为中间体的成盐离子导致,因此在小浓度下进样NaCl进行了排查,对比结果如图13所示。NaCl保留时间和较大杂质峰保留时间一致,可能为相应对离子。此外,在NQAD系统使用的甲酸铵流动相体系下,中间体2和3得到了分离。[/align][align=left][/align][align=center][img=,690,455]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251551127175_1278_2222981_3.jpg!w690x455.jpg[/img][/align][align=center]图13 不同样品NQAD比较图[/align][img=,637,239]http://ng1.17img.cn/bbsfiles/images/2018/01/201801251551124165_6276_2222981_3.jpg!w637x239.jpg[/img][align=left][/align][align=left]综上所述,使用大阪曹達PC HILIC S5 4.6 mm i.d. × 250 mm色谱柱可完成左卡尼汀中间体及成品的保留,在紫外检测条件下获得较好峰形结果,能够与死时间附近的杂质峰取得良好分离。[/align]
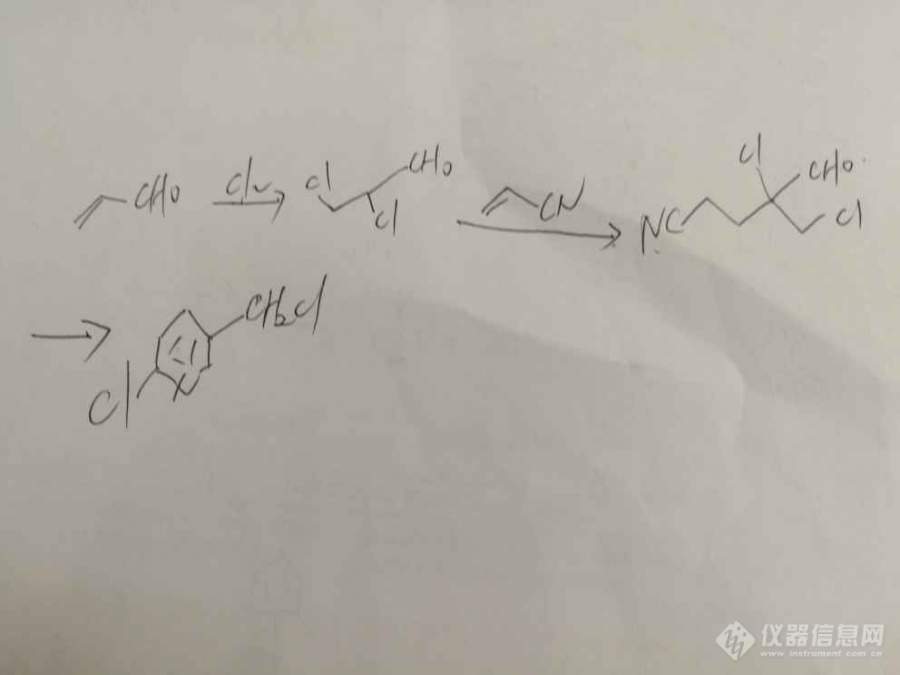
[color=#444444]正在做吡虫啉中间体CCMP,按照如下步骤合成。但反应需要用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]监控,有没有人能提供第一步和第二步的[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测方法?不胜感激!![/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907011027506739_3504_1801607_3.jpg!w690x517.jpg[/img][/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP